

Risdiplam in Patients Previously Treated with Other Therapies for Spinal Muscular Atrophy: An Interim Analysis from the JEWELFISH Study
- PMID: 36780114
- PMCID: PMC9924181
- DOI: 10.1007/s40120-023-00444-1
Risdiplam in Patients Previously Treated with Other Therapies for Spinal Muscular Atrophy: An Interim Analysis from the JEWELFISH Study
Erratum in
-
Correction to: Risdiplam in Patients Previously Treated with Other Therapies for Spinal Muscular Atrophy: An Interim Analysis from the JEWELFISH Study.Neurol Ther. 2023 Oct;12(5):1799-1801. doi: 10.1007/s40120-023-00503-7. Neurol Ther. 2023. PMID: 37395990 Free PMC article. No abstract available.
Abstract
Introduction: Risdiplam is a survival of motor neuron 2 (SMN2) splicing modifier for the treatment of patients with spinal muscular atrophy (SMA). The JEWELFISH study (NCT03032172) was designed to assess the safety, tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of risdiplam in previously treated pediatric and adult patients with types 1-3 SMA. Here, an analysis was performed after all patients had received at least 1 year of treatment with risdiplam.
Methods: Patients with a confirmed diagnosis of 5q-autosomal recessive SMA between the ages of 6 months and 60 years were eligible for enrollment. Patients were previously enrolled in the MOONFISH study (NCT02240355) with splicing modifier RG7800 or treated with olesoxime, nusinersen, or onasemnogene abeparvovec. The primary objectives of the JEWELFISH study were to evaluate the safety and tolerability of risdiplam and investigate the PK after 2 years of treatment.
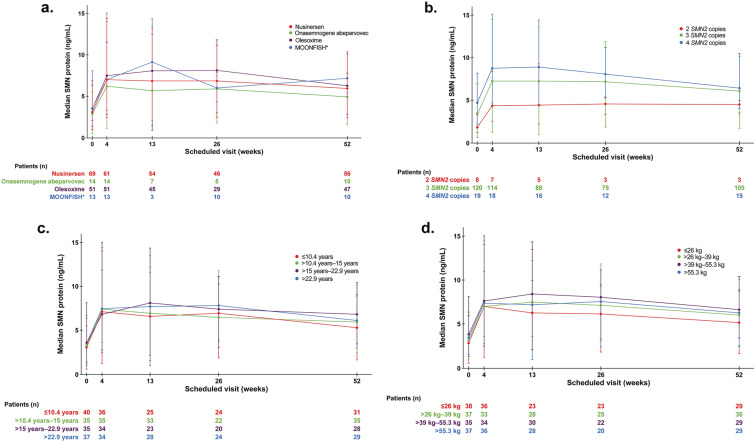
Results: A total of 174 patients enrolled: MOONFISH study (n = 13), olesoxime (n = 71 patients), nusinersen (n = 76), onasemnogene abeparvovec (n = 14). Most patients (78%) had three SMN2 copies. The median age and weight of patients at enrollment was 14.0 years (1-60 years) and 39.1 kg (9.2-108.9 kg), respectively. About 63% of patients aged 2-60 years had a baseline total score of less than 10 on the Hammersmith Functional Motor Scale-Expanded and 83% had scoliosis. The most common adverse event (AE) was upper respiratory tract infection and pyrexia (30 patients each; 17%). Pneumonia (four patients; 2%) was the most frequently reported serious AE (SAE). The rates of AEs and SAEs per 100 patient-years were lower in the second 6-month period compared with the first. An increase in SMN protein was observed in blood after risdiplam treatment and was comparable across all ages and body weight quartiles.
Conclusions: The safety and PD of risdiplam in patients who were previously treated were consistent with those of treatment-naïve patients.
Keywords: Evrysdi; Pharmacodynamics; Risdiplam; Safety; Spinal muscular atrophy.
© 2023. The Author(s).
Conflict of interest statement
Birgit Jaber, Carmen Martin, Francis Warren, Heidemarie Kletzl, Ksenija Gorni, Renata S. Scalco, and Kathryn R. Wagner are employees and shareholders of F. Hoffmann-La Roche Ltd, Basel, Switzerland. At the time of the study, Imogen Carruthers was an employee of F. Hoffmann-La Roche Ltd, Basel, Switzerland and is now an employee of CS Genetics. Claudia A. Chiriboga has taken part in advisory boards for AveXis/Novartis, Biogen, PTC Therapeutics, Genentech, and F. Hoffmann-La Roche, received educational speaker fees from Biogen, F. Hoffmann-La Roche and Genentech and received research support grants from AveXis/Novartis, Biogen, F. Hoffmann-La Roche, and the National Institutes of Health. Claudio Bruno has received honoraria for scientific advisory boards from Biogen, Novartis, F. Hoffmann-La Roche, and Sarepta Therapeutics, and has received research support grants from Biogen. Tina Duong has received honoraria for scientific advisory boards or consultancy from Biogen, Novartis, F. Hoffmann-La Roche, Genentech, Pfizer, Sarepta Therapeutics, Audentes, Astellas, and Dyne. Dirk Fischer has received honoraria for scientific advisory boards or consultancy from F. Hoffmann-La Roche. Eugenio Mercuri has received honoraria for scientific advisory boards and educational speaker fees from Sarepta Therapeutics, Santhera, PTC Therapeutics, Pfizer, F. Hoffmann-La Roche, Biogen, Avexis, Novartis, Scholar Rock, and Cytokinetics, and has received research support grants from Biogen. Janbernd Kirschner has received honoraria for scientific advisory boards from Biogen, Novartis, PTC Therapeutics, Pfizer, F. Hoffmann-La Roche, Sarepta Therapeutics and reports grants from Biogen, Novartis, and F. Hoffmann-La Roche. Anna Kostera-Pruszczyk has received compensation for participation at symposia, lectures, and scientific advisory boards from Biogen, F. Hoffmann-La Roche, AveXis/Novartis and PTC Therapeutics. She has also received institutional grant support from Biogen and is an investigator in SMA trials sponsored by F. Hoffmann-La Roche. Francesco Muntoni has received honoraria for scientific advisory boards from Novartis Gene Therapies, Inc., Biogen, PTC Therapeutics, Sarepta Therapeutics, F. Hoffmann-La Roche and reports grants and personal fees from Novartis Gene Therapies, Inc., Biogen, Sarepta Therapeutics and F. Hoffmann-La Roche. FM is member of the Pfizer Rare Disease Advisory Board and of Dyne Therapeutic SAB.
Figures
References
-
- Munsat T. Workshop report: international SMA collaboration. Neuromuscul Disord. 1991;1:81. doi: 10.1016/0960-8966(91)90052-T. - DOI
-
- New three-year data for Roche’s Evrysdi (risdiplam) show long-term improvements in survival and motor milestones in babies with type 1 spinal muscular atrophy (SMA) 2022. https://www.roche.com/media/releases/med-cor-2022-04-29. Accessed Nov 2022.
LinkOut - more resources
Full Text Sources
