

ALK Amplification and Rearrangements Are Recurrent Targetable Events in Congenital and Adult Glioblastoma
- PMID: 36780194
- PMCID: PMC10363218
- DOI: 10.1158/1078-0432.CCR-21-3521
ALK Amplification and Rearrangements Are Recurrent Targetable Events in Congenital and Adult Glioblastoma
Abstract
Purpose: Anaplastic lymphoma kinase (ALK) aberrations have been identified in pediatric-type infant gliomas, but their occurrence across age groups, functional effects, and treatment response has not been broadly established.
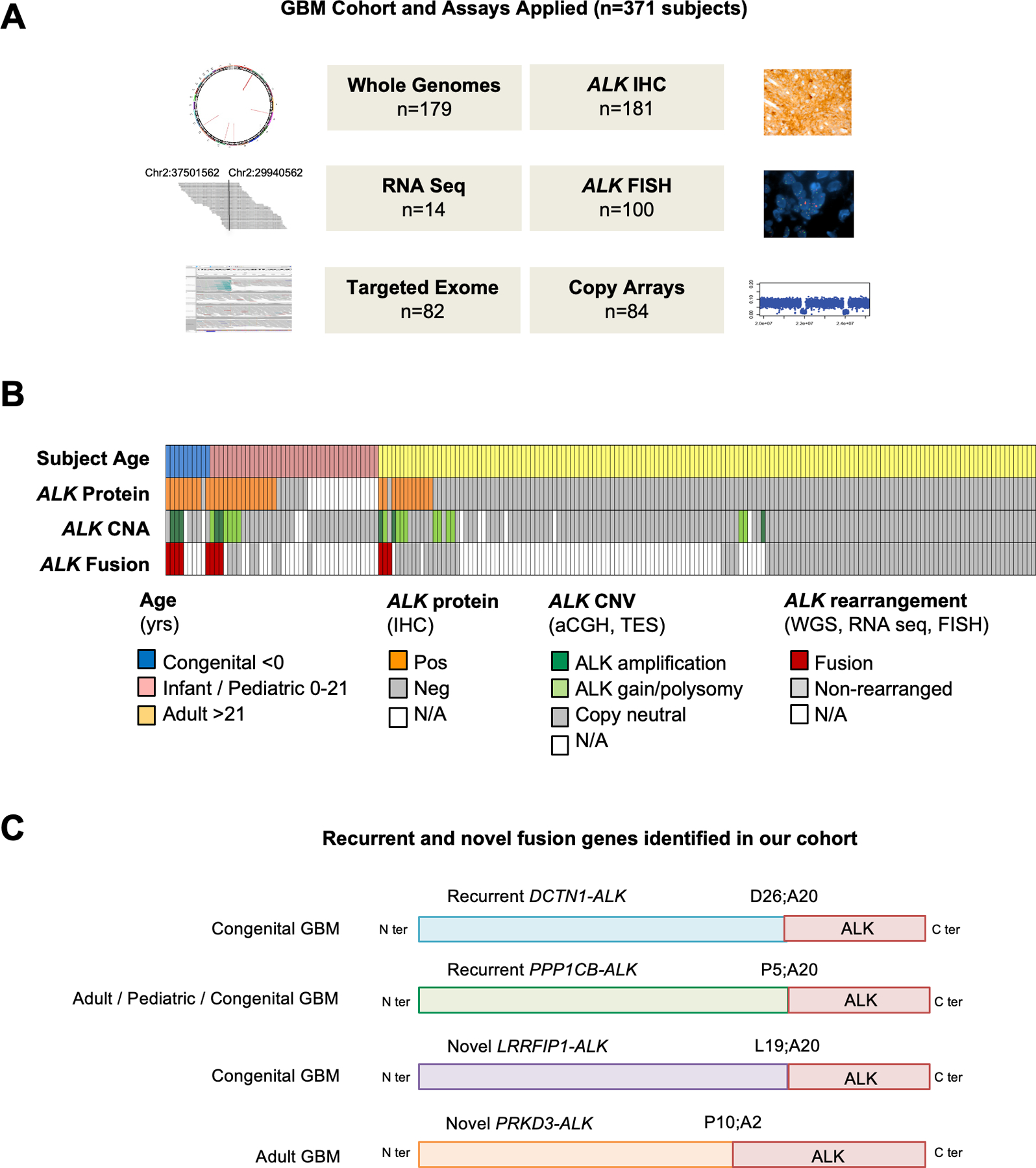
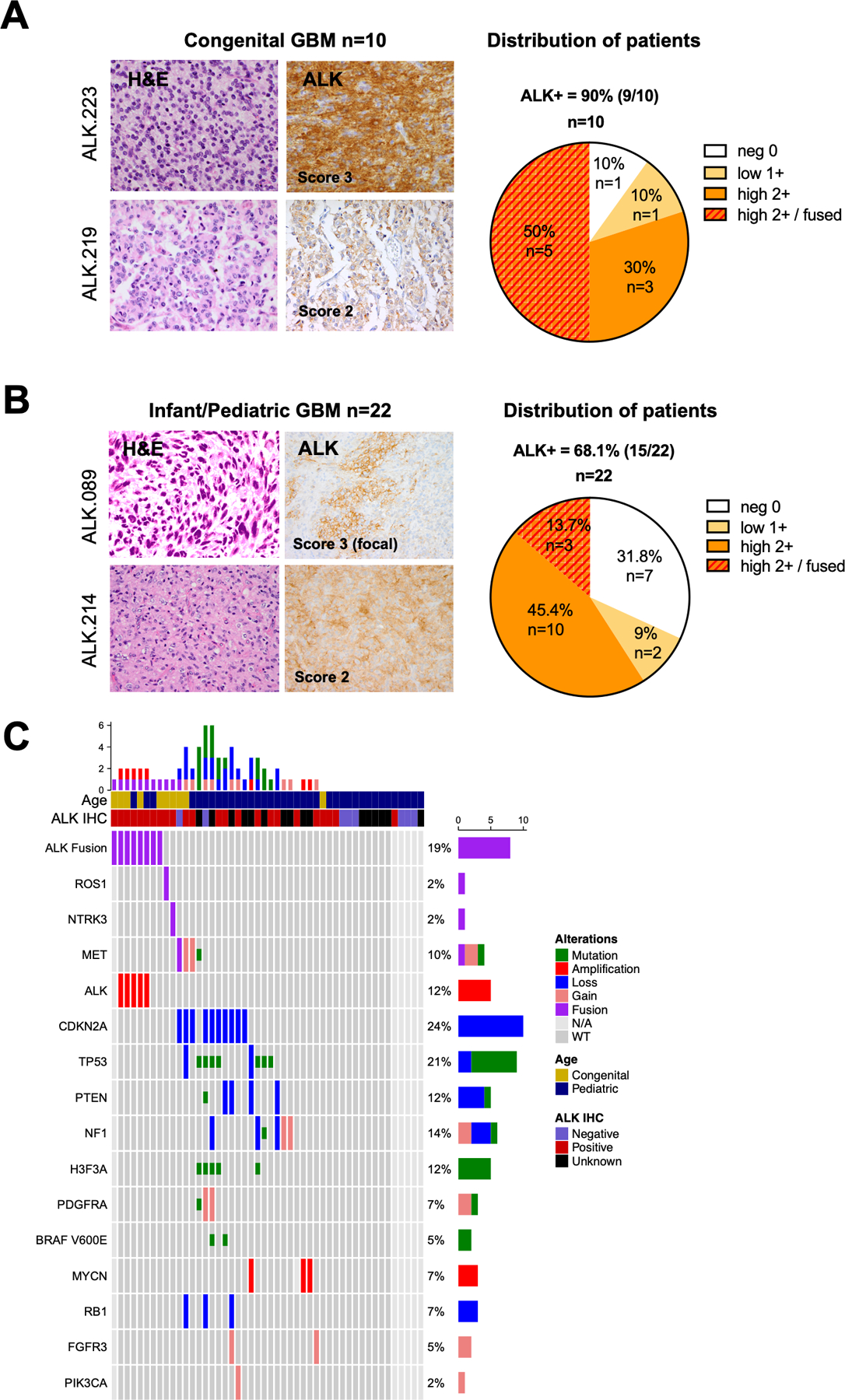
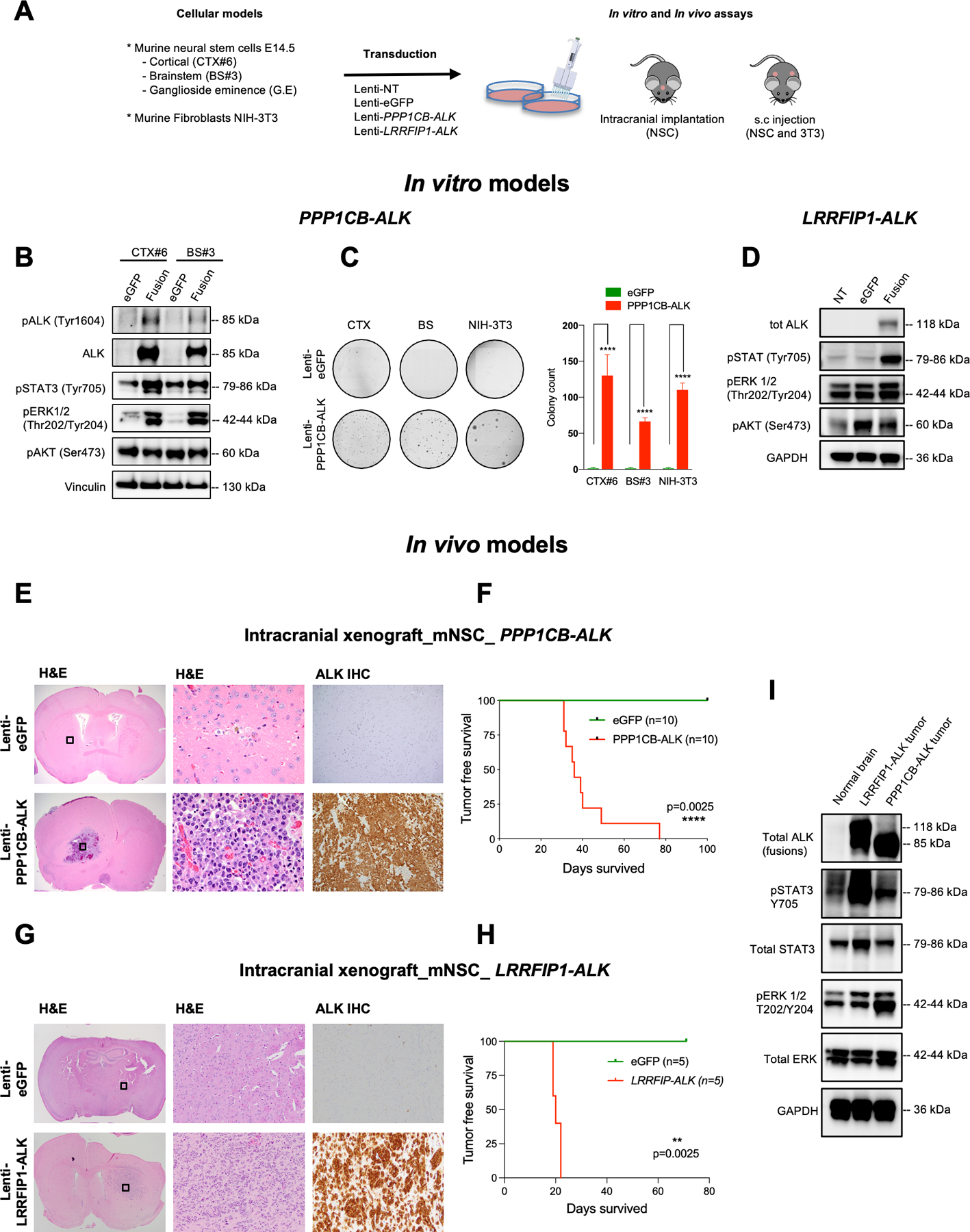
Experimental design: We performed a comprehensive analysis of ALK expression and genomic aberrations in both newly generated and retrospective data from 371 glioblastomas (156 adult, 205 infant/pediatric, and 10 congenital) with in vitro and in vivo validation of aberrations.
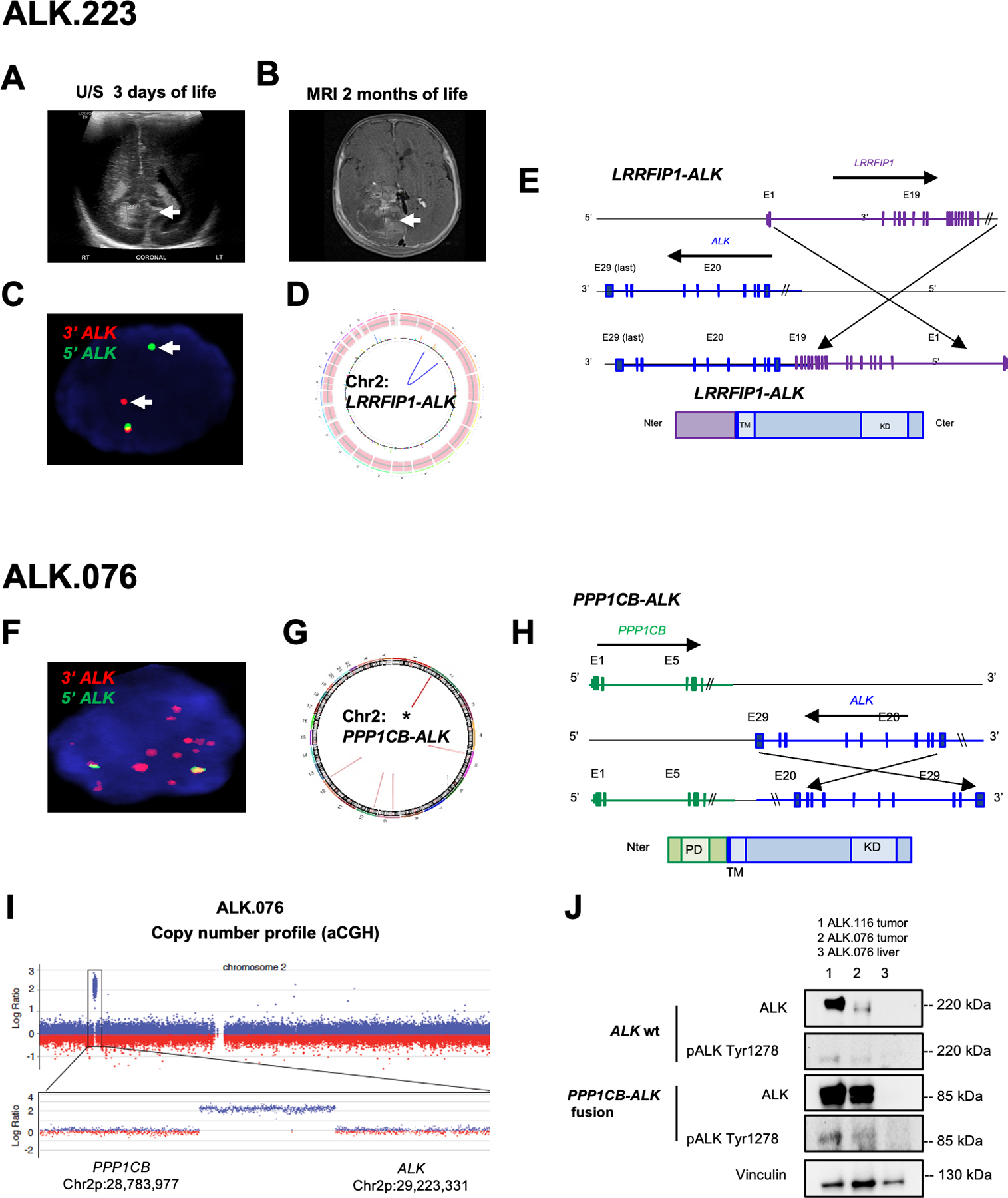
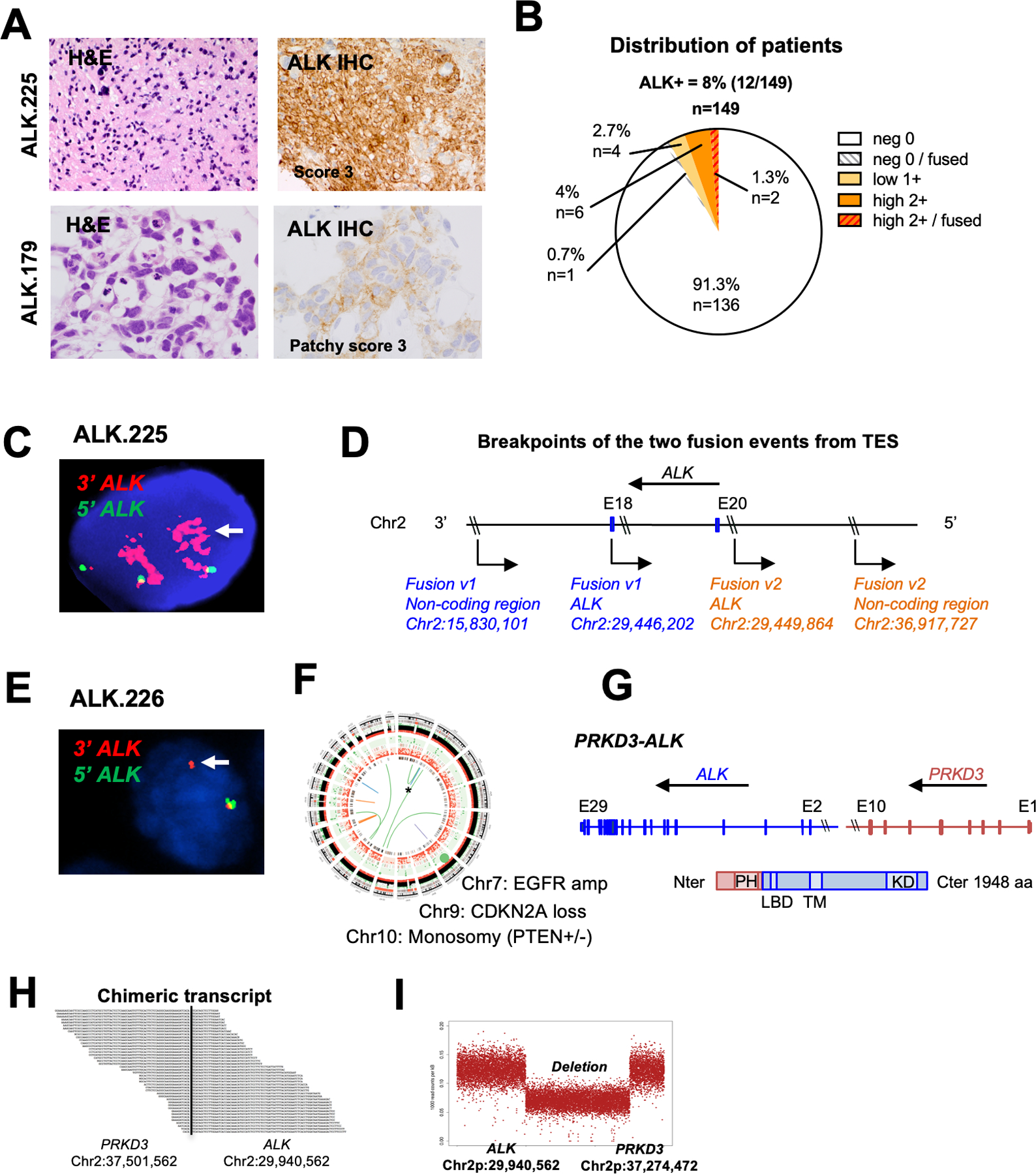
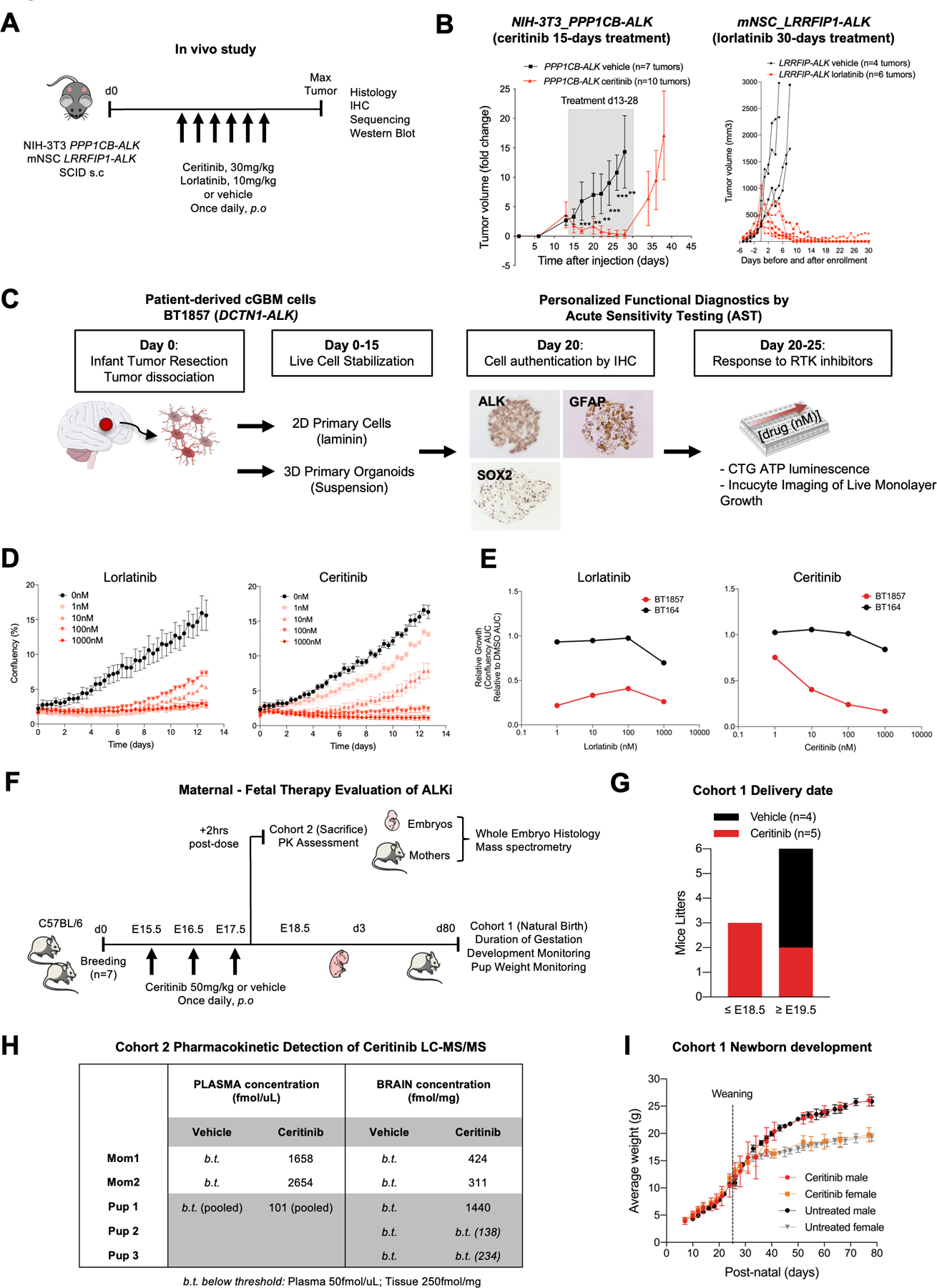
Results: ALK aberrations at the protein or genomic level were detected in 12% of gliomas (45/371) in a wide age range (0-80 years). Recurrent as well as novel ALK fusions (LRRFIP1-ALK, DCTN1-ALK, PRKD3-ALK) were present in 50% (5/10) of congenital/infant, 1.4% (3/205) of pediatric, and 1.9% (3/156) of adult GBMs. ALK fusions were present as the only candidate driver in congenital/infant GBMs and were sometimes focally amplified. In contrast, adult ALK fusions co-occurred with other oncogenic drivers. No activating ALK mutations were identified in any age group. Novel and recurrent ALK rearrangements promoted STAT3 and ERK1/2 pathways and transformation in vitro and in vivo. ALK-fused GBM cellular and mouse models were responsive to ALK inhibitors, including in patient cells derived from a congenital GBM. Relevant to the treatment of infant gliomas, we showed that ALK protein appears minimally expressed in the forebrain at perinatal stages, and no gross effects on perinatal brain development were seen in pregnant mice treated with the ALK inhibitor ceritinib.
Conclusions: These findings support use of brain-penetrant ALK inhibitors in clinical trials across infant, pediatric, and adult GBMs. See related commentary by Mack and Bertrand, p. 2567.
©2023 American Association for Cancer Research.
Figures
Comment in
-
A Molecular Blueprint to Targeting ALK Gene Fusions in Glioblastoma.Clin Cancer Res. 2023 Jul 14;29(14):2567-2569. doi: 10.1158/1078-0432.CCR-23-0472. Clin Cancer Res. 2023. PMID: 37260294
References
-
- El-Ayadi M, Ansari M, Kühnöl CD, Bendel A, Sturm D, Pietsch T, et al. Occurrence of high-grade glioma in Noonan syndrome: Report of two cases. Pediatr Blood Cancer 2019;66:e27625. - PubMed
-
- Korostyshevskaya AM, Savelov AA, Papusha LI, Druy AE, Yarnykh VL. Congenital medulloblastoma: Fetal and postnatal longitudinal observation with quantitative MRI. Clin Imaging 2018;52:172–6. - PubMed
-
- Kostadinov S, de la Monte S. A Case of Congenital Brainstem Oligodendroglioma: Pathology Findings and Review of the Literature. Case Rep Neurol Med [Internet] 2017. [cited 2019 Jul 15];2017. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5549497/ - PMC - PubMed
-
- Anestis D, Tsitsopoulos P, Ble C, Tsitouras V, Tsonidis C. Congenital Glioblastoma Multiforme: An Unusual and Challenging Tumor. Neuropediatrics 2017;48:403–12. - PubMed
-
- Hallberg B, Palmer RH. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat Rev Cancer 2013;13:685–700. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
