

Structural basis of efficacy-driven ligand selectivity at GPCRs
- PMID: 36782010
- PMCID: PMC10299909
- DOI: 10.1038/s41589-022-01247-5
Structural basis of efficacy-driven ligand selectivity at GPCRs
Erratum in
-
Author Correction: Structural basis of efficacy-driven ligand selectivity at GPCRs.Nat Chem Biol. 2023 Apr;19(4):529. doi: 10.1038/s41589-023-01297-3. Nat Chem Biol. 2023. PMID: 36879062 Free PMC article. No abstract available.
Abstract
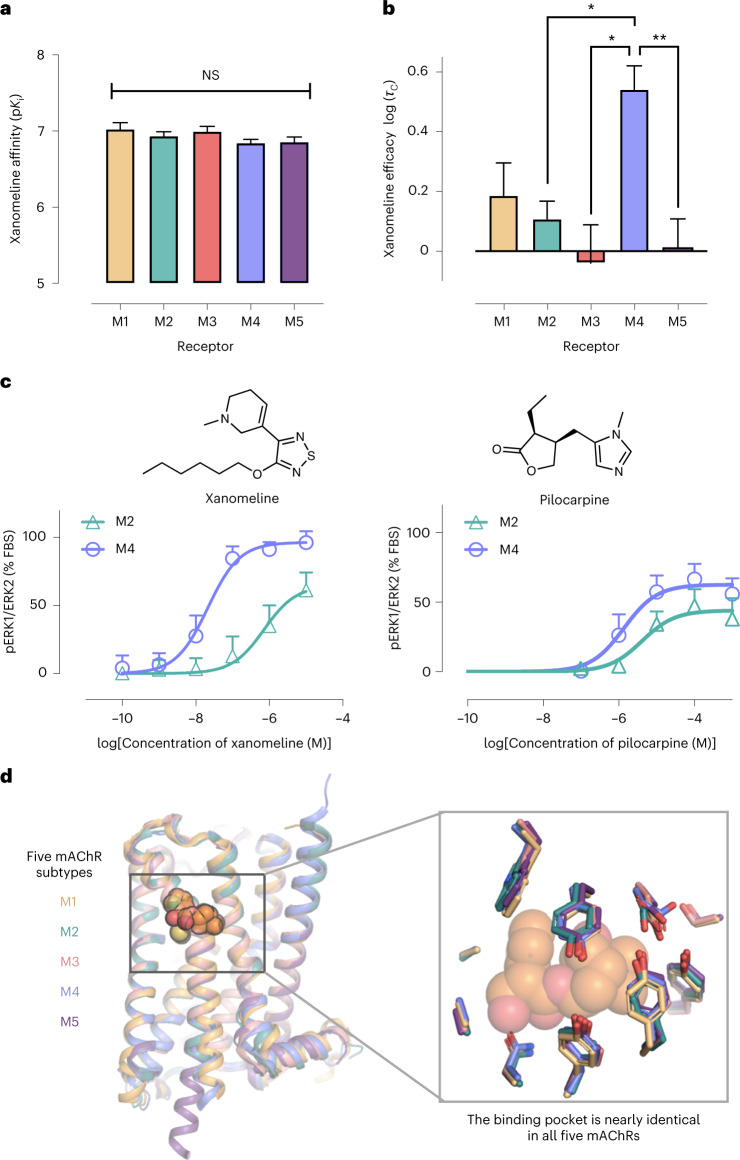
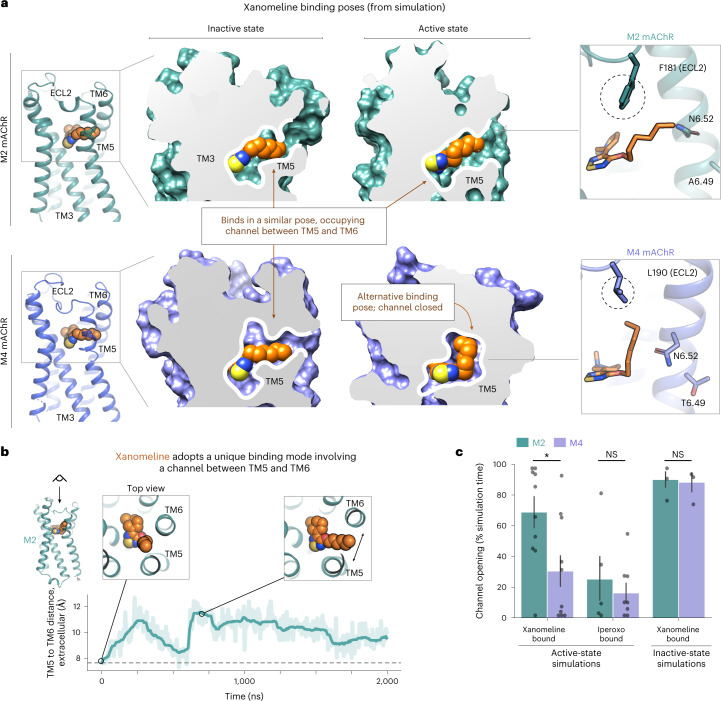
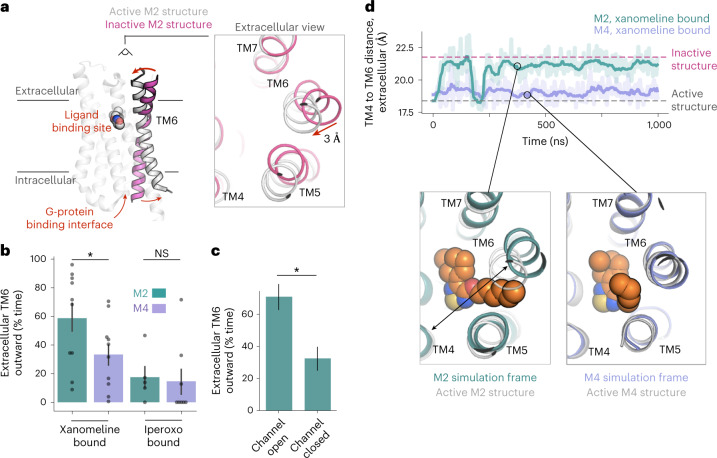
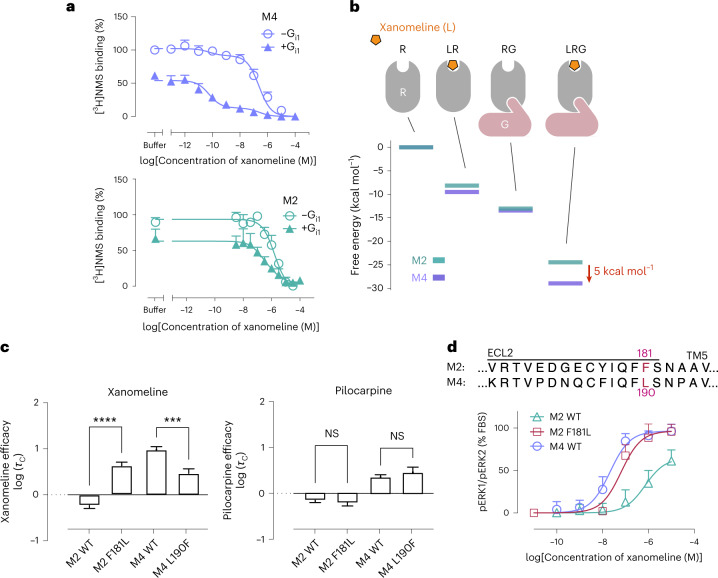
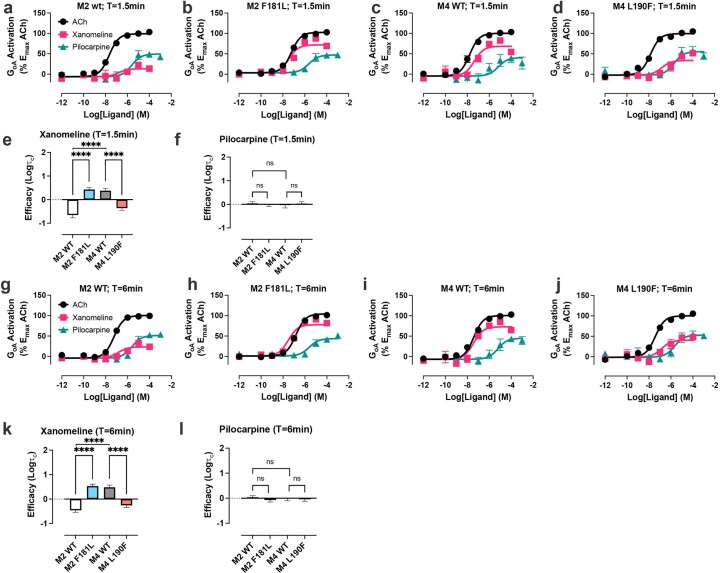
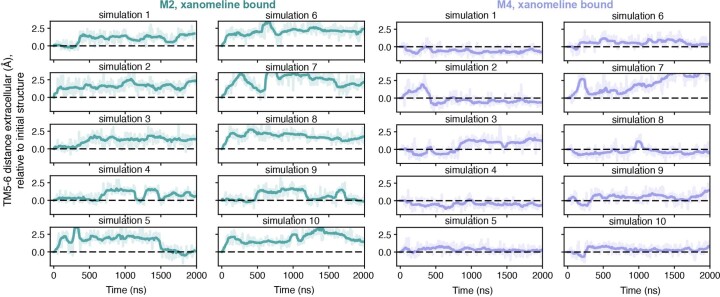
A drug's selectivity for target receptors is essential to its therapeutic utility, but achieving selectivity between similar receptors is challenging. The serendipitous discovery of ligands that stimulate target receptors more strongly than closely related receptors, despite binding with similar affinities, suggests a solution. The molecular mechanism of such 'efficacy-driven selectivity' has remained unclear, however, hindering design of such ligands. Here, using atomic-level simulations, we reveal the structural basis for the efficacy-driven selectivity of a long-studied clinical drug candidate, xanomeline, between closely related muscarinic acetylcholine receptors (mAChRs). Xanomeline's binding mode is similar across mAChRs in their inactive states but differs between mAChRs in their active states, with divergent effects on active-state stability. We validate this mechanism experimentally and use it to design ligands with altered efficacy-driven selectivity. Our results suggest strategies for the rational design of ligands that achieve efficacy-driven selectivity for many pharmaceutically important G-protein-coupled receptors.
© 2023. The Author(s).
Conflict of interest statement
C.C.F. and S.M.P. are employees of and hold equity in Karuna Therapeutics. A.C. and P.M.S. are co-founders of Septerna, Inc. A.C., P.M.S. and R.O.D. hold equity in Septerna, Inc.
Figures





Similar articles
-
Biased Profile of Xanomeline at the Recombinant Human M4 Muscarinic Acetylcholine Receptor.ACS Chem Neurosci. 2022 Apr 20;13(8):1206-1218. doi: 10.1021/acschemneuro.1c00827. Epub 2022 Apr 5. ACS Chem Neurosci. 2022. PMID: 35380782
-
Entropy drives the ligand recognition in G-protein-coupled receptor subtypes.Proc Natl Acad Sci U S A. 2024 Jul 23;121(30):e2401091121. doi: 10.1073/pnas.2401091121. Epub 2024 Jul 18. Proc Natl Acad Sci U S A. 2024. PMID: 39024109 Free PMC article.
-
Striatal, Hippocampal, and Cortical Networks Are Differentially Responsive to the M4- and M1-Muscarinic Acetylcholine Receptor Mediated Effects of Xanomeline.ACS Chem Neurosci. 2019 Mar 20;10(3):1753-1764. doi: 10.1021/acschemneuro.8b00625. Epub 2018 Dec 11. ACS Chem Neurosci. 2019. PMID: 30480428
-
Classics in Chemical Neuroscience: Xanomeline.ACS Chem Neurosci. 2017 Mar 15;8(3):435-443. doi: 10.1021/acschemneuro.7b00001. Epub 2017 Feb 13. ACS Chem Neurosci. 2017. PMID: 28141924 Review.
-
Structure-function studies of muscarinic acetylcholine receptors.Handb Exp Pharmacol. 2012;(208):29-48. doi: 10.1007/978-3-642-23274-9_2. Handb Exp Pharmacol. 2012. PMID: 22222693 Review.
Cited by
-
Structure-guided design of partial agonists at an opioid receptor.Nat Commun. 2025 Mar 13;16(1):2518. doi: 10.1038/s41467-025-57734-5. Nat Commun. 2025. PMID: 40082451 Free PMC article.
-
Mapping the space of protein binding sites with sequence-based protein language models.Bioinformatics. 2025 Jun 2;41(6):btaf284. doi: 10.1093/bioinformatics/btaf284. Bioinformatics. 2025. PMID: 40576205 Free PMC article.
-
Novel Xanomeline-Containing Bitopic Ligands of Muscarinic Acetylcholine Receptors: Design, Synthesis and FRET Investigation.Molecules. 2023 Mar 6;28(5):2407. doi: 10.3390/molecules28052407. Molecules. 2023. PMID: 36903650 Free PMC article.
-
CXC Chemokine Ligand 12 Facilitates Gi Protein Binding to CXC Chemokine Receptor 4 by Stabilizing Packing of the Proline-Isoleucine-Phenylalanine Motif: Insights from Automated Path Searching.J Am Chem Soc. 2025 Mar 26;147(12):10129-10138. doi: 10.1021/jacs.4c14293. Epub 2025 Mar 17. J Am Chem Soc. 2025. PMID: 40096846 Free PMC article.
-
GPCR-IPL score: multilevel featurization of GPCR-ligand interaction patterns and prediction of ligand functions from selectivity to biased activation.Brief Bioinform. 2024 Jan 22;25(2):bbae105. doi: 10.1093/bib/bbae105. Brief Bioinform. 2024. PMID: 38517694 Free PMC article.
References
-
- Campillos M, Kuhn M, Gavin AC, Jensen LJ, Bork P. Drug target identification using side-effect similarity. Science. 2008;321:263–266. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
