

Whole genome sequencing for USH2A-associated disease reveals several pathogenic deep-intronic variants that are amenable to splice correction
- PMID: 36785559
- PMCID: PMC9918427
- DOI: 10.1016/j.xhgg.2023.100181
Whole genome sequencing for USH2A-associated disease reveals several pathogenic deep-intronic variants that are amenable to splice correction
Abstract
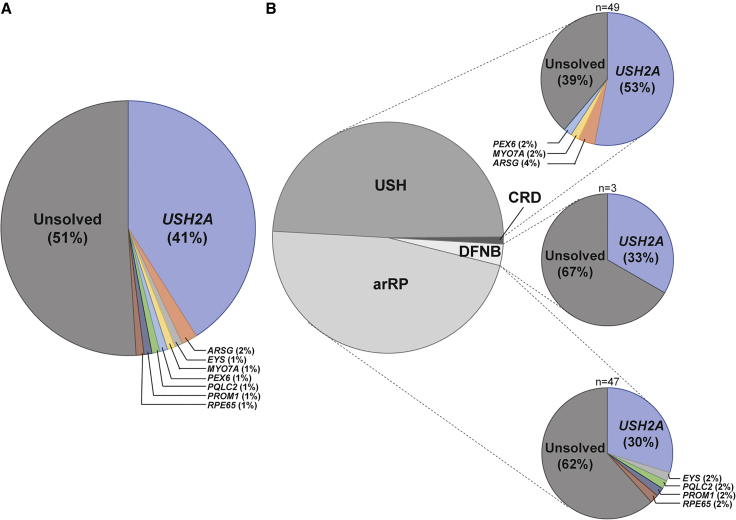
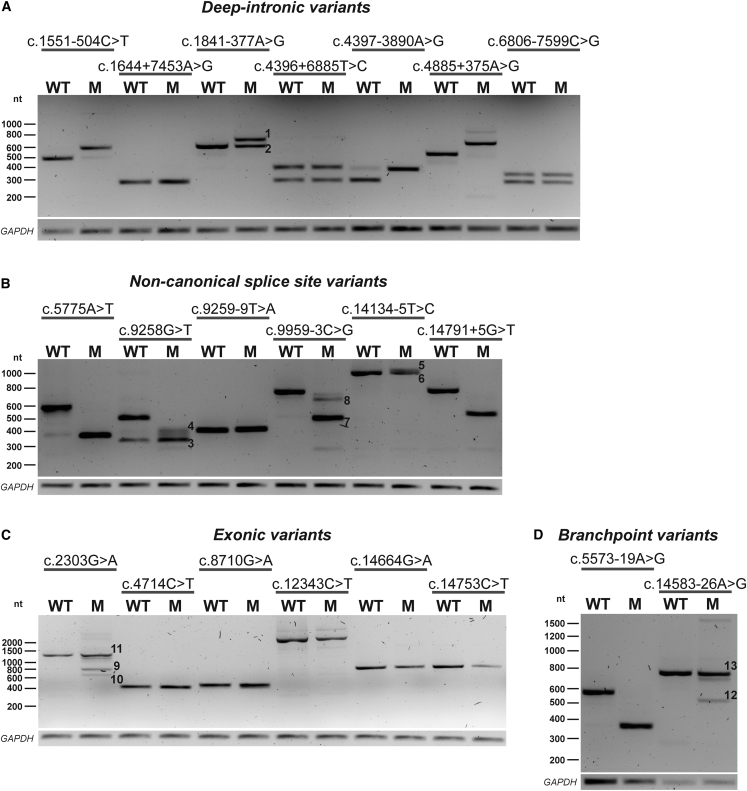
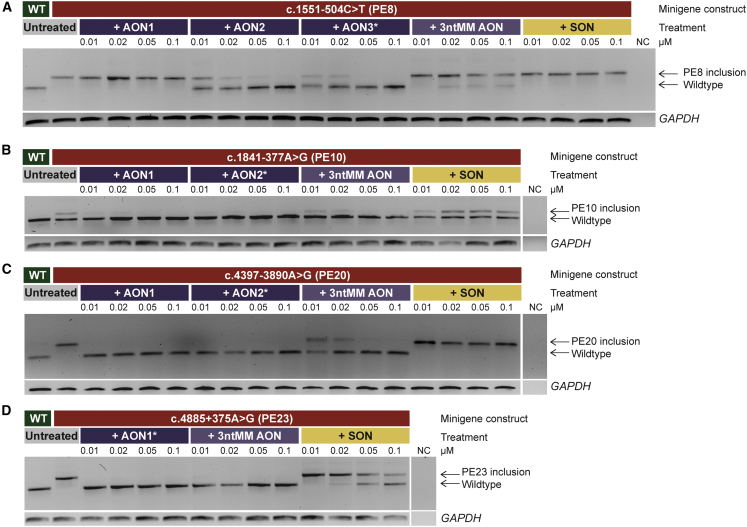
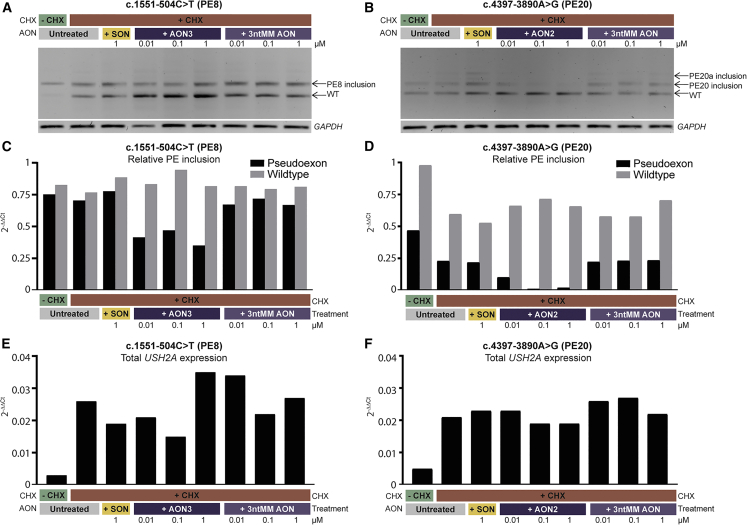
A significant number of individuals with a rare disorder such as Usher syndrome (USH) and (non-)syndromic autosomal recessive retinitis pigmentosa (arRP) remain genetically unexplained. Therefore, we assessed subjects suspected of USH2A-associated disease and no or mono-allelic USH2A variants using whole genome sequencing (WGS) followed by an improved pipeline for variant interpretation to provide a conclusive diagnosis. One hundred subjects were screened using WGS to identify causative variants in USH2A or other USH/arRP-associated genes. In addition to the existing variant interpretation pipeline, a particular focus was put on assessing splice-affecting properties of variants, both in silico and in vitro. Also structural variants were extensively addressed. For variants resulting in pseudoexon inclusion, we designed and evaluated antisense oligonucleotides (AONs) using minigene splice assays and patient-derived photoreceptor precursor cells. Biallelic variants were identified in 49 of 100 subjects, including novel splice-affecting variants and structural variants, in USH2A or arRP/USH-associated genes. Thirteen variants were shown to affect USH2A pre-mRNA splicing, including four deep-intronic USH2A variants resulting in pseudoexon inclusion, which could be corrected upon AON treatment. We have shown that WGS, combined with a thorough variant interpretation pipeline focused on assessing pre-mRNA splicing defects and structural variants, is a powerful method to provide subjects with a rare genetic condition, a (likely) conclusive genetic diagnosis. This is essential for the development of future personalized treatments and for patients to be eligible for such treatments.
Keywords: USH2A; Usher syndrome; antisense oligonucleotides; minigene splice assay; photoreceptor precursor cells; pseudoexon; retinitis pigmentosa; splicing; usherin; whole genome sequencing.
© 2023 The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Vaché C., Besnard T., le Berre P., García-García G., Baux D., Larrieu L., Abadie C., Blanchet C., Bolz H.J., Millan J., et al. Usher syndrome type 2 caused by activation of an USH2A pseudoexon: implications for diagnosis and therapy. Hum. Mutat. 2012;33:104–108. doi: 10.1002/humu.21634. - DOI - PubMed
-
- Baux D., Vaché C., Blanchet C., Willems M., Baudoin C., Moclyn M., Faugère V., Touraine R., Isidor B., Dupin-Deguine D., et al. Combined genetic approaches yield a 48% diagnostic rate in a large cohort of French hearing-impaired patients. Sci. Rep. 2017;7:16783. doi: 10.1038/s41598-017-16846-9. - DOI - PMC - PubMed
-
- Carss K.J., Arno G., Erwood M., Stephens J., Sanchis-Juan A., Hull S., Megy K., Grozeva D., Dewhurst E., Malka S., et al. Comprehensive rare variant analysis via whole-genome sequencing to determine the molecular pathology of inherited retinal disease. Am. J. Hum. Genet. 2017;100:75–90. doi: 10.1016/j.ajhg.2016.12.003. - DOI - PMC - PubMed
-
- de Bruijn S.E., Fiorentino A., Ottaviani D., Fanucchi S., Melo U.S., Corral-Serrano J.C., Mulders T., Georgiou M., Rivolta C., Pontikos N., et al. Structural variants create new topological-associated domains and ectopic retinal enhancer-gene contact in dominant retinitis pigmentosa. Am. J. Hum. Genet. 2020;107:802–814. doi: 10.1016/j.ajhg.2020.09.002. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
