

Complet+: a computationally scalable method to improve completeness of large-scale protein sequence clustering
- PMID: 36785708
- PMCID: PMC9921987
- DOI: 10.7717/peerj.14779
Complet+: a computationally scalable method to improve completeness of large-scale protein sequence clustering
Abstract
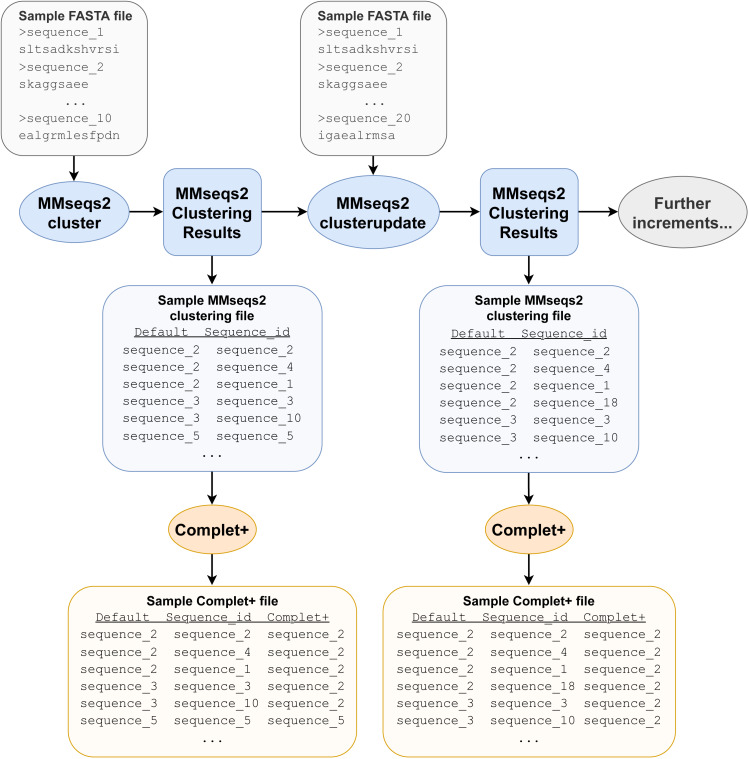
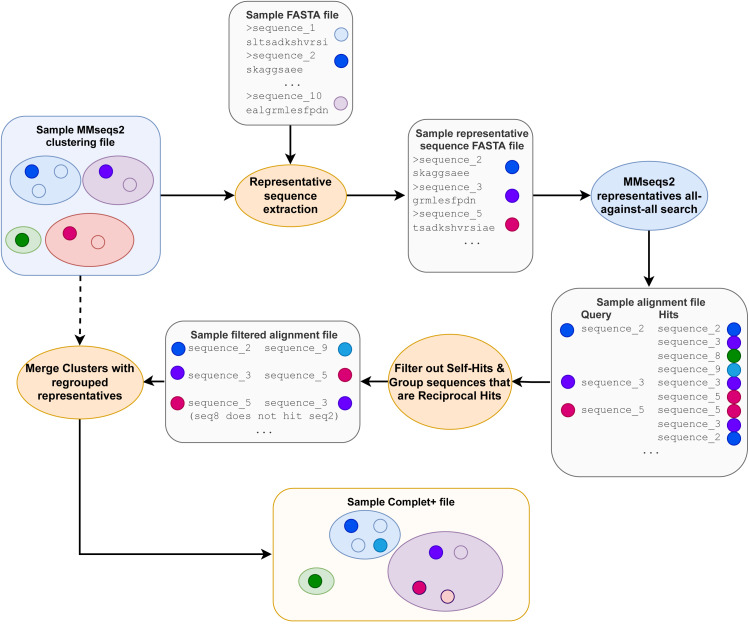
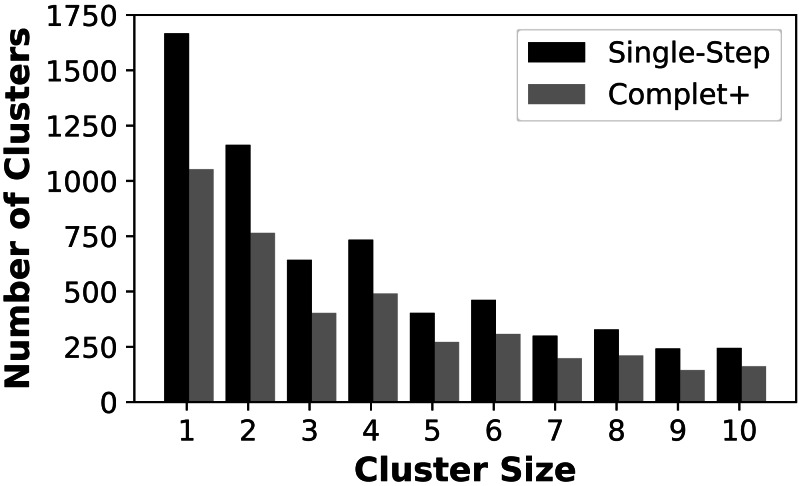
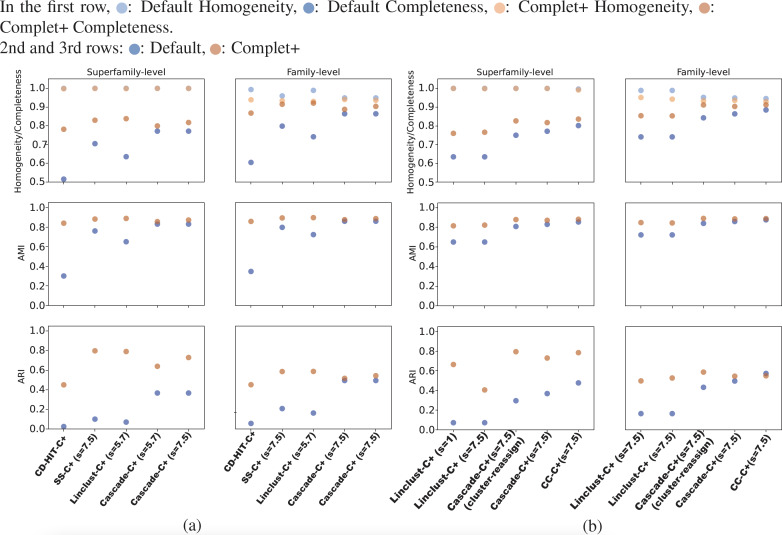
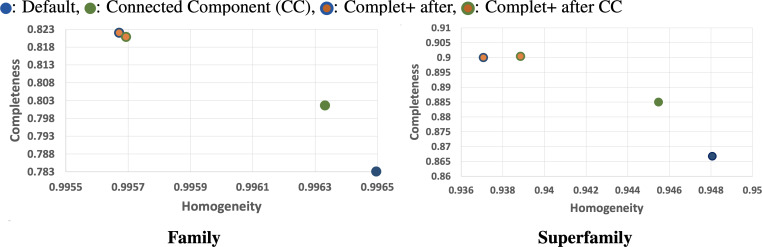
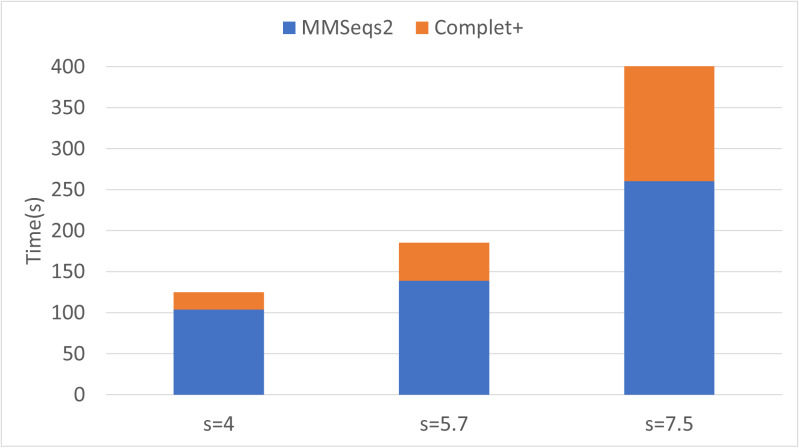
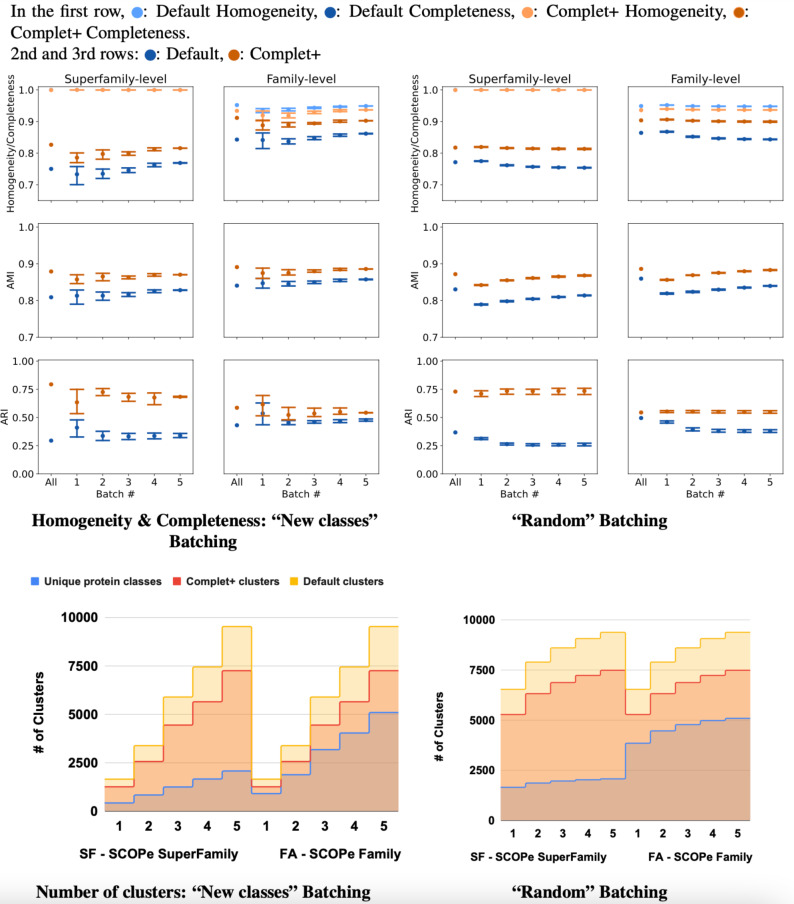
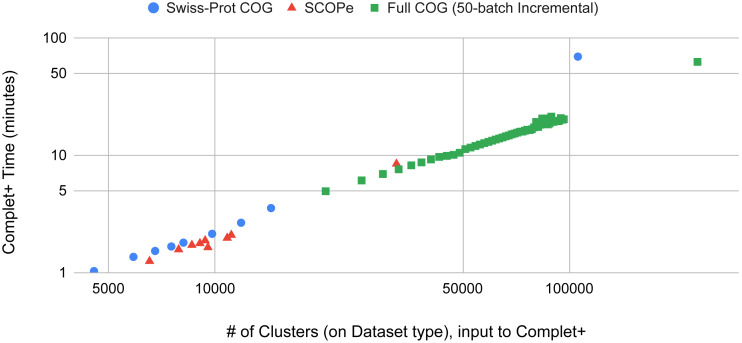
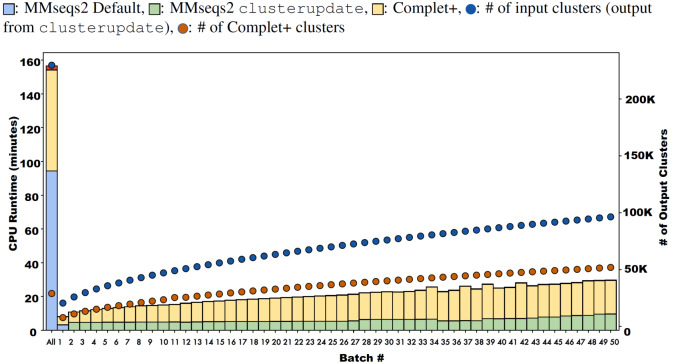
A major challenge for clustering algorithms is to balance the trade-off between homogeneity, i.e., the degree to which an individual cluster includes only related sequences, and completeness, the degree to which related sequences are broken up into multiple clusters. Most algorithms are conservative in grouping sequences with other sequences. Remote homologs may fail to be clustered together and instead form unnecessarily distinct clusters. The resulting clusters have high homogeneity but completeness that is too low. We propose Complet+, a computationally scalable post-processing method to increase the completeness of clusters without an undue cost in homogeneity. Complet+ proves to effectively merge closely-related clusters of protein that have verified structural relationships in the SCOPe classification scheme, improving the completeness of clustering results at little cost to homogeneity. Applying Complet+ to clusters obtained using MMseqs2's clusterupdate achieves an increased V-measure of 0.09 and 0.05 at the SCOPe superfamily and family levels, respectively. Complet+ also creates more biologically representative clusters, as shown by a substantial increase in Adjusted Mutual Information (AMI) and Adjusted Rand Index (ARI) metrics when comparing predicted clusters to biological classifications. Complet+ similarly improves clustering metrics when applied to other methods, such as CD-HIT and linclust. Finally, we show that Complet+ runtime scales linearly with respect to the number of clusters being post-processed on a COG dataset of over 3 million sequences. Code and supplementary information is available on Github: https://github.com/EESI/Complet-Plus.
Keywords: Homology; Protein clustering; Protein families.
©2023 Nguyen et al.
Conflict of interest statement
The authors declare there are no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
