Molecular pathways that drive diabetic kidney disease
- PMID: 36787250
- PMCID: PMC9927939
- DOI: 10.1172/JCI165654
Molecular pathways that drive diabetic kidney disease
Abstract
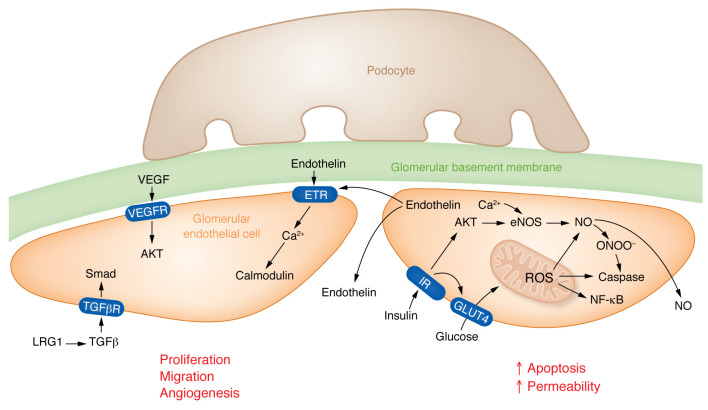
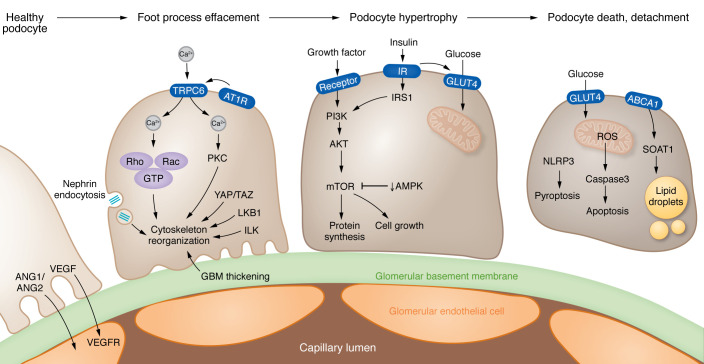
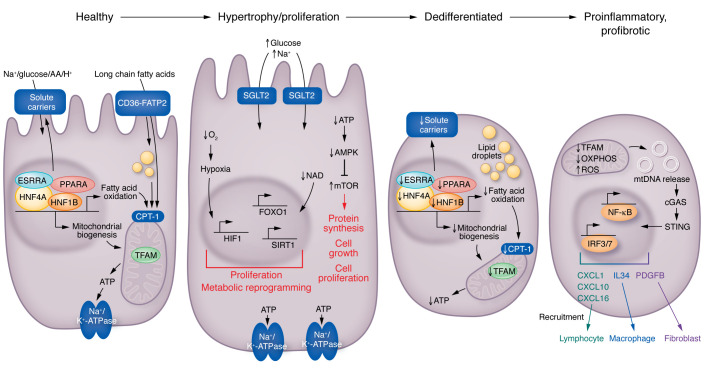
Kidney disease is a major driver of mortality among patients with diabetes and diabetic kidney disease (DKD) is responsible for close to half of all chronic kidney disease cases. DKD usually develops in a genetically susceptible individual as a result of poor metabolic (glycemic) control. Molecular and genetic studies indicate the key role of podocytes and endothelial cells in driving albuminuria and early kidney disease in diabetes. Proximal tubule changes show a strong association with the glomerular filtration rate. Hyperglycemia represents a key cellular stress in the kidney by altering cellular metabolism in endothelial cells and podocytes and by imposing an excess workload requiring energy and oxygen for proximal tubule cells. Changes in metabolism induce early adaptive cellular hypertrophy and reorganization of the actin cytoskeleton. Later, mitochondrial defects contribute to increased oxidative stress and activation of inflammatory pathways, causing progressive kidney function decline and fibrosis. Blockade of the renin-angiotensin system or the sodium-glucose cotransporter is associated with cellular protection and slowing kidney function decline. Newly identified molecular pathways could provide the basis for the development of much-needed novel therapeutics.
Conflict of interest statement
Figures