

Accelerated SARS-CoV-2 intrahost evolution leading to distinct genotypes during chronic infection
- PMID: 36791724
- PMCID: PMC9906997
- DOI: 10.1016/j.xcrm.2023.100943
Accelerated SARS-CoV-2 intrahost evolution leading to distinct genotypes during chronic infection
Abstract
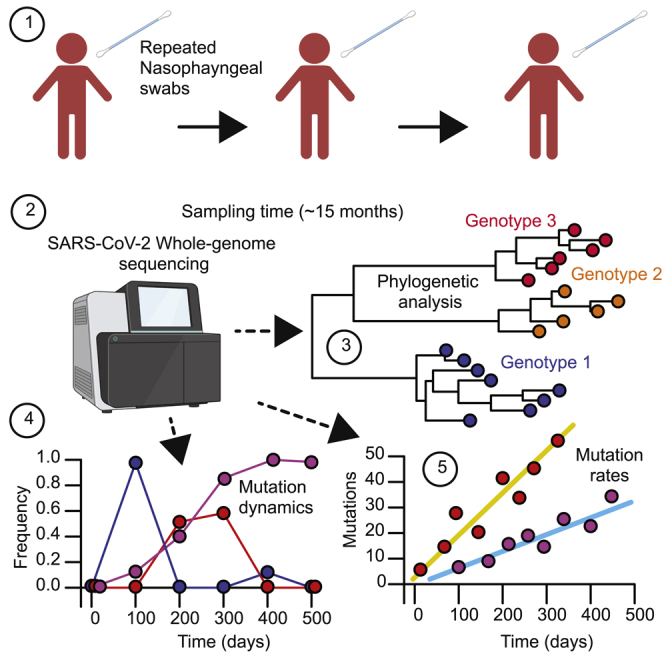
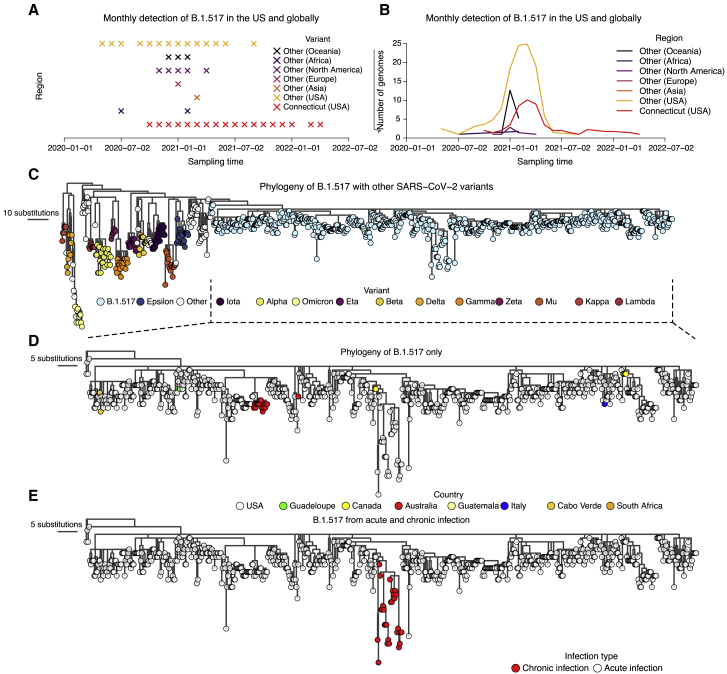
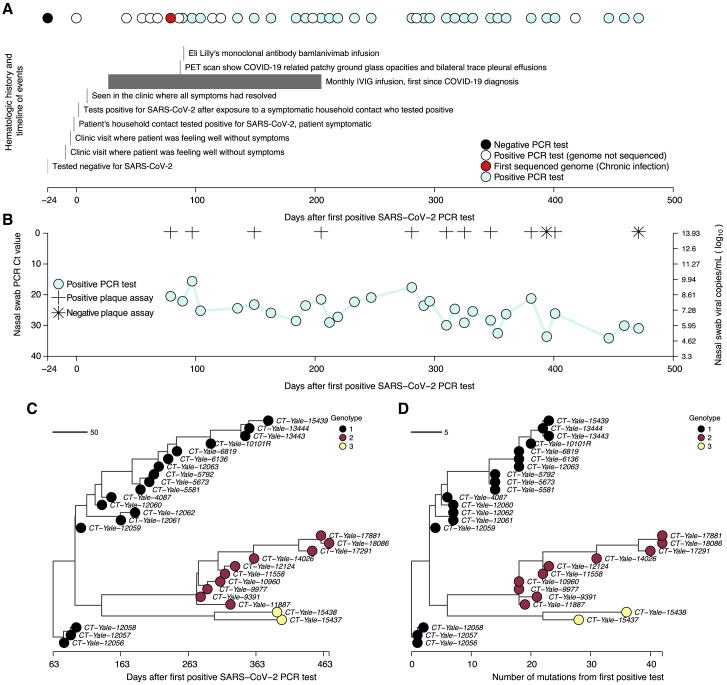
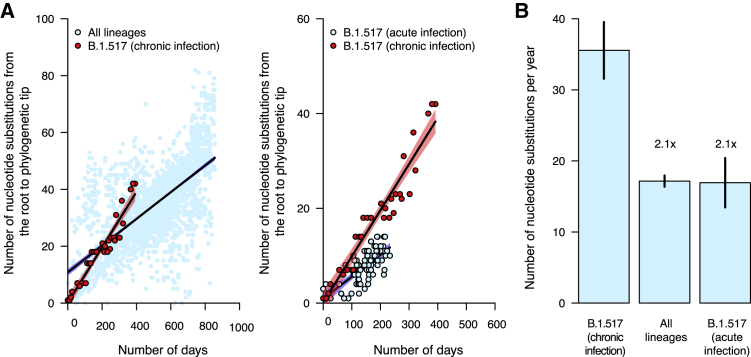
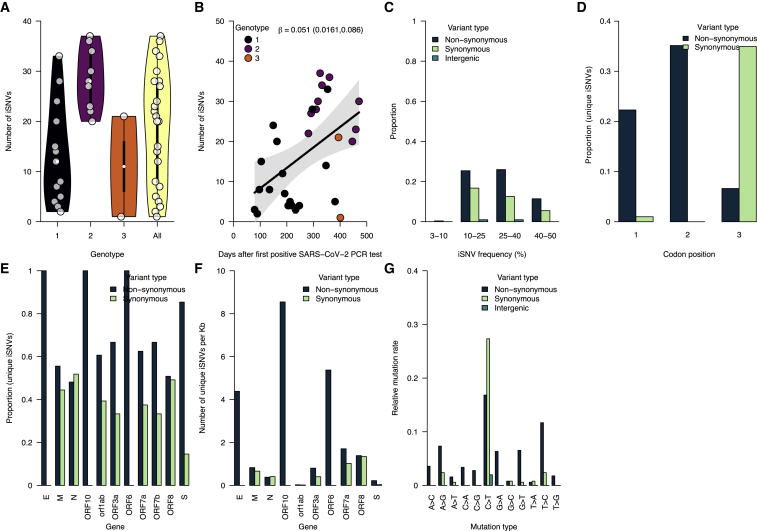
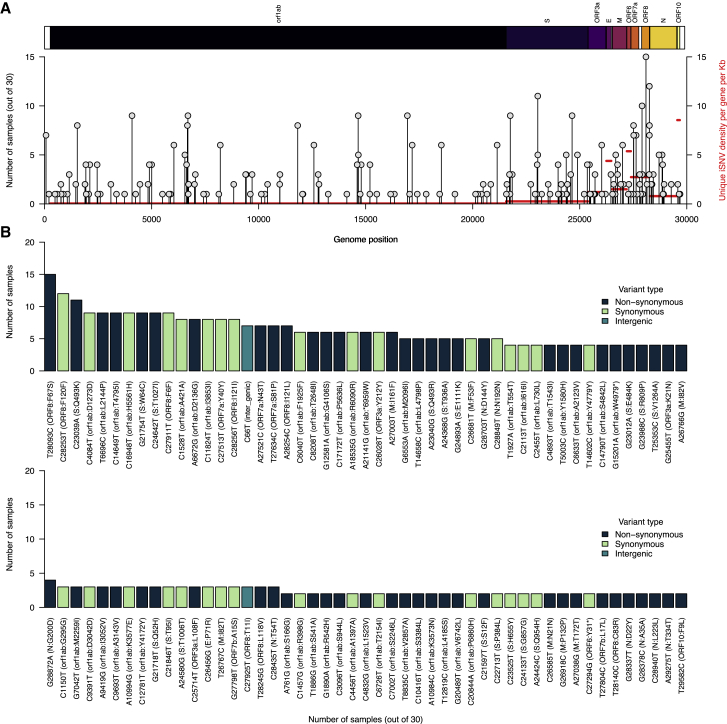
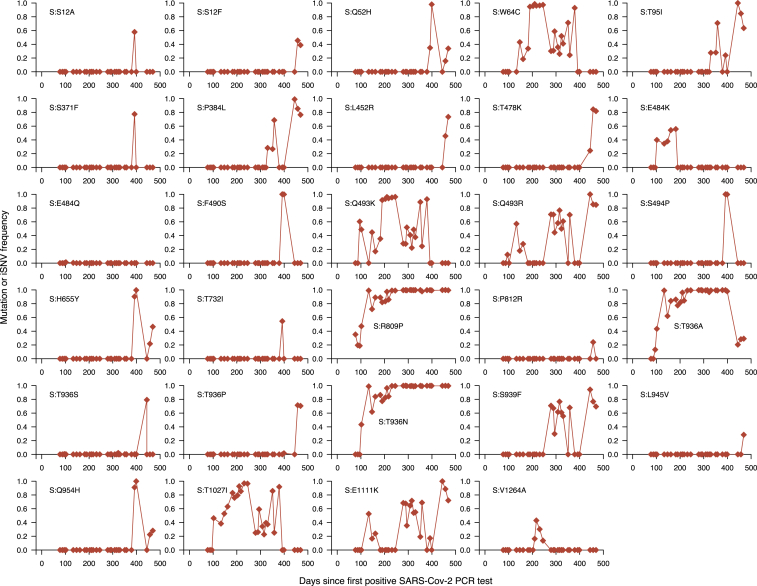
The chronic infection hypothesis for novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variant emergence is increasingly gaining credence following the appearance of Omicron. Here, we investigate intrahost evolution and genetic diversity of lineage B.1.517 during a SARS-CoV-2 chronic infection lasting for 471 days (and still ongoing) with consistently recovered infectious virus and high viral genome copies. During the infection, we find an accelerated virus evolutionary rate translating to 35 nucleotide substitutions per year, approximately 2-fold higher than the global SARS-CoV-2 evolutionary rate. This intrahost evolution results in the emergence and persistence of at least three genetically distinct genotypes, suggesting the establishment of spatially structured viral populations continually reseeding different genotypes into the nasopharynx. Finally, we track the temporal dynamics of genetic diversity to identify advantageous mutations and highlight hallmark changes for chronic infection. Our findings demonstrate that untreated chronic infections accelerate SARS-CoV-2 evolution, providing an opportunity for the emergence of genetically divergent variants.
Keywords: COVID-19 vaccines; SARS-CoV-2; chronic infection; epidemiology; genomic surveillance; immunocompromised individual; intrahost evolution; intrahost genotypes; mutation dynamics; variant emergence.
Copyright © 2023 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests N.D.G. is a consultant for Tempus Labs and the National Basketball Association for work related to COVID-19 but is outside the submitted work. The University of North Carolina is pursuing intellectual property protection for Primer ID sequencing, and R.I.S. has received nominal royalties from licensing.
Figures
Update of
-
Accelerated SARS-CoV-2 intrahost evolution leading to distinct genotypes during chronic infection.medRxiv [Preprint]. 2022 Jul 2:2022.06.29.22276868. doi: 10.1101/2022.06.29.22276868. medRxiv. 2022. Update in: Cell Rep Med. 2023 Feb 21;4(2):100943. doi: 10.1016/j.xcrm.2023.100943. PMID: 35794895 Free PMC article. Updated. Preprint.
References
-
- Takashita E., Kinoshita N., Yamayoshi S., Sakai-Tagawa Y., Fujisaki S., Ito M., Iwatsuki-Horimoto K., Chiba S., Halfmann P., Nagai H., et al. Efficacy of antibodies and antiviral drugs against Covid-19 Omicron variant. N. Engl. J. Med. 2022;386:995–998. doi: 10.1056/NEJMc2119407. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
