

A NPAS4-NuA4 complex couples synaptic activity to DNA repair
- PMID: 36792830
- PMCID: PMC9946837
- DOI: 10.1038/s41586-023-05711-7
A NPAS4-NuA4 complex couples synaptic activity to DNA repair
Abstract
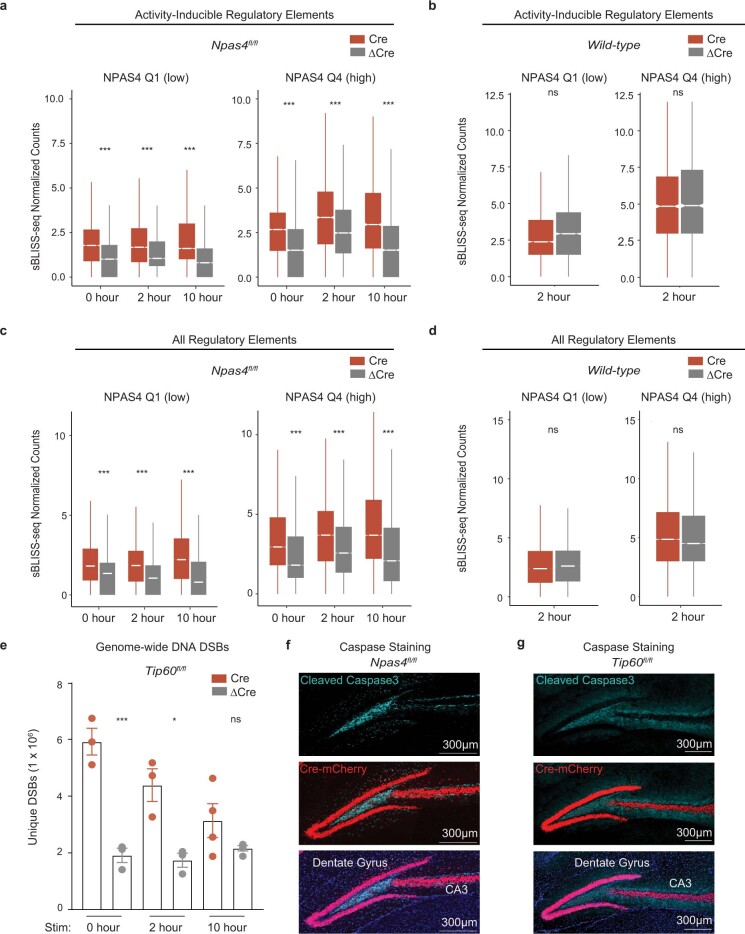
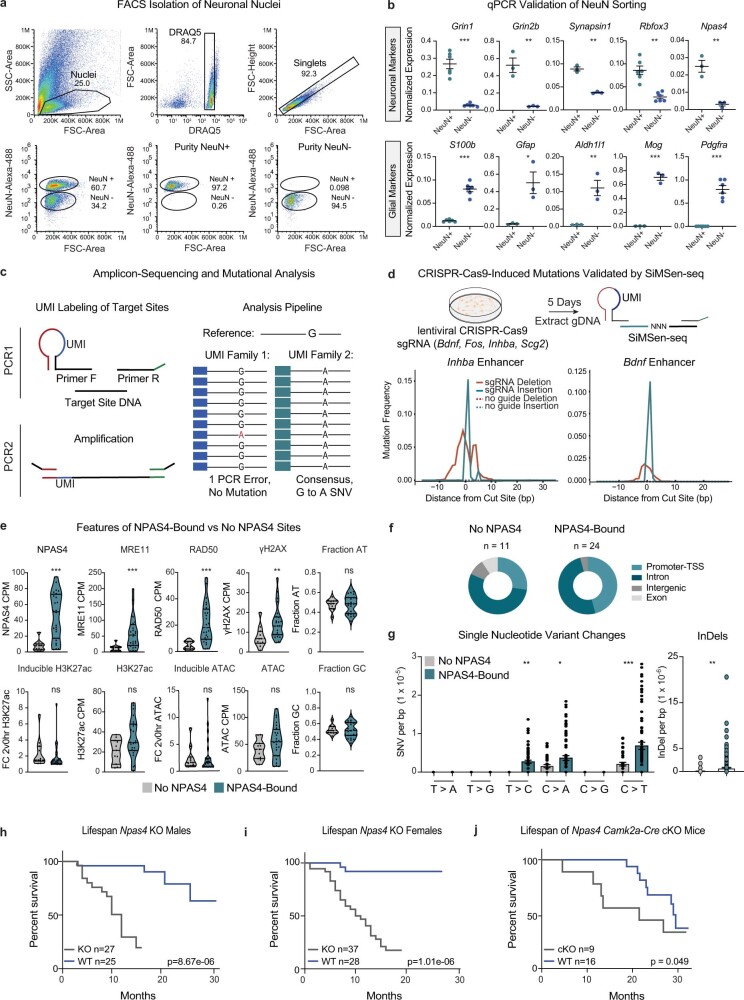
Neuronal activity is crucial for adaptive circuit remodelling but poses an inherent risk to the stability of the genome across the long lifespan of postmitotic neurons1-5. Whether neurons have acquired specialized genome protection mechanisms that enable them to withstand decades of potentially damaging stimuli during periods of heightened activity is unknown. Here we identify an activity-dependent DNA repair mechanism in which a new form of the NuA4-TIP60 chromatin modifier assembles in activated neurons around the inducible, neuronal-specific transcription factor NPAS4. We purify this complex from the brain and demonstrate its functions in eliciting activity-dependent changes to neuronal transcriptomes and circuitry. By characterizing the landscape of activity-induced DNA double-strand breaks in the brain, we show that NPAS4-NuA4 binds to recurrently damaged regulatory elements and recruits additional DNA repair machinery to stimulate their repair. Gene regulatory elements bound by NPAS4-NuA4 are partially protected against age-dependent accumulation of somatic mutations. Impaired NPAS4-NuA4 signalling leads to a cascade of cellular defects, including dysregulated activity-dependent transcriptional responses, loss of control over neuronal inhibition and genome instability, which all culminate to reduce organismal lifespan. In addition, mutations in several components of the NuA4 complex are reported to lead to neurodevelopmental and autism spectrum disorders. Together, these findings identify a neuronal-specific complex that couples neuronal activity directly to genome preservation, the disruption of which may contribute to developmental disorders, neurodegeneration and ageing.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
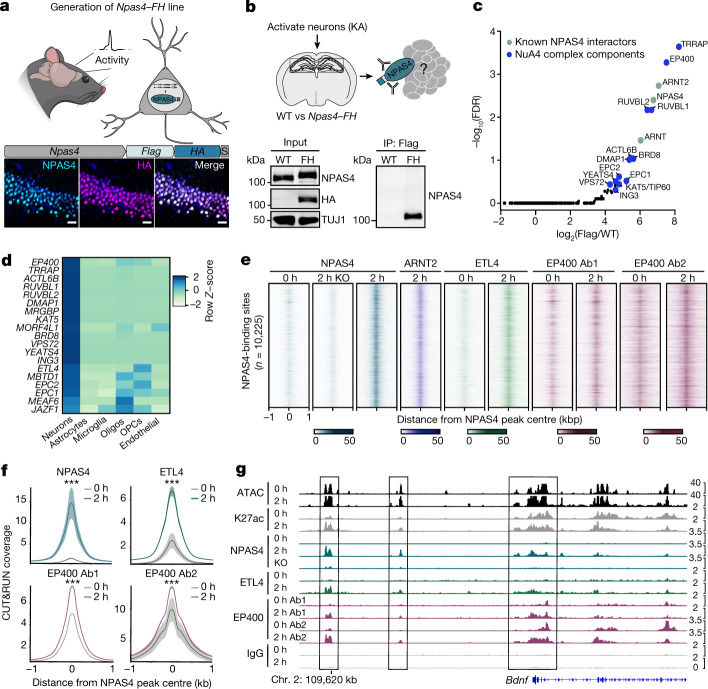
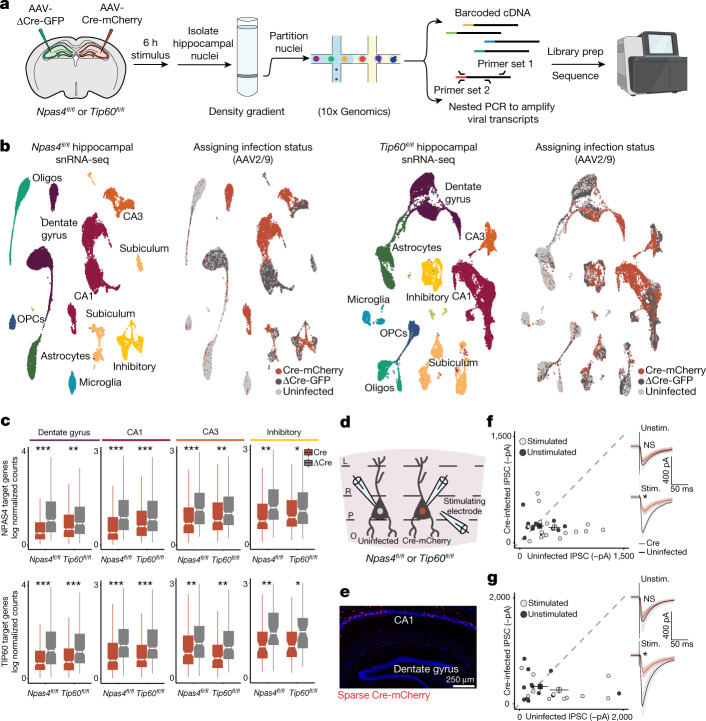
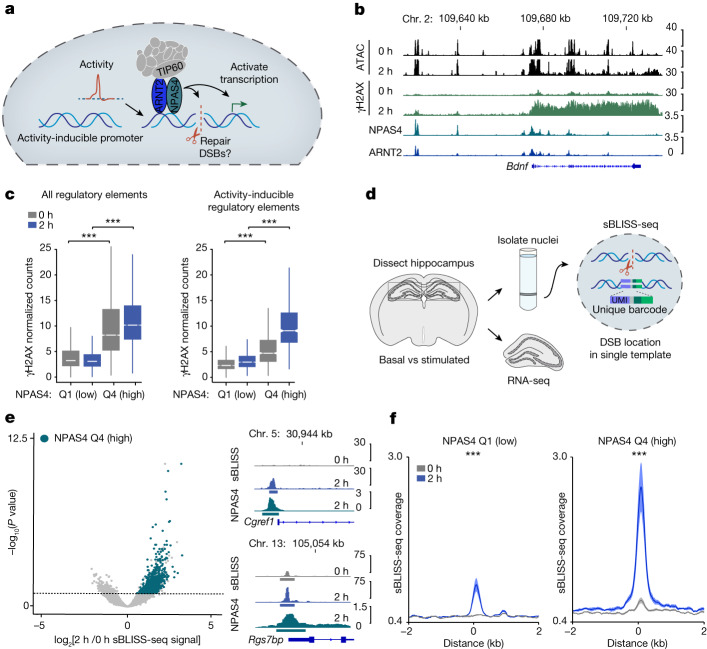
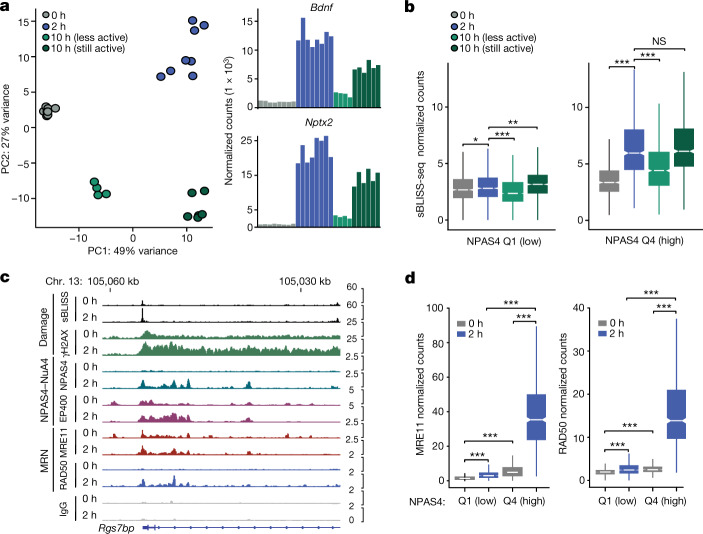
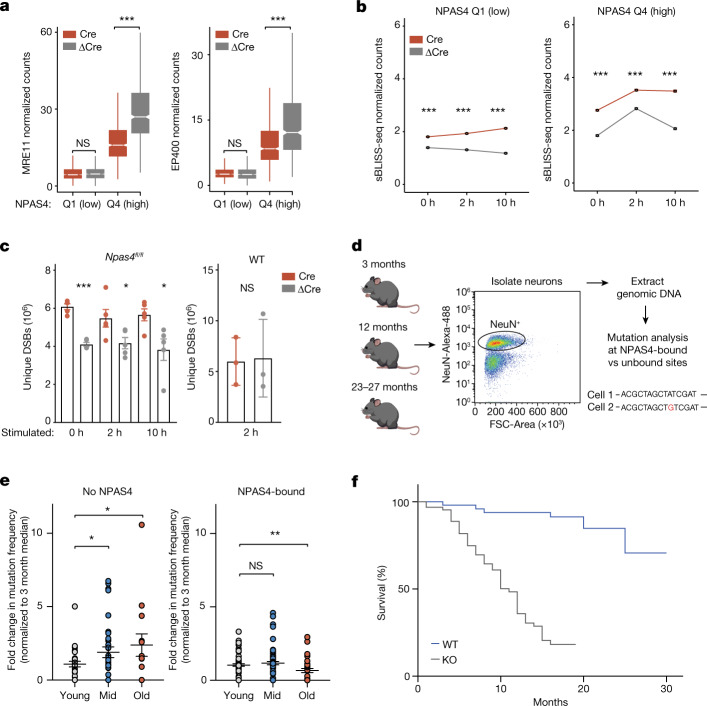
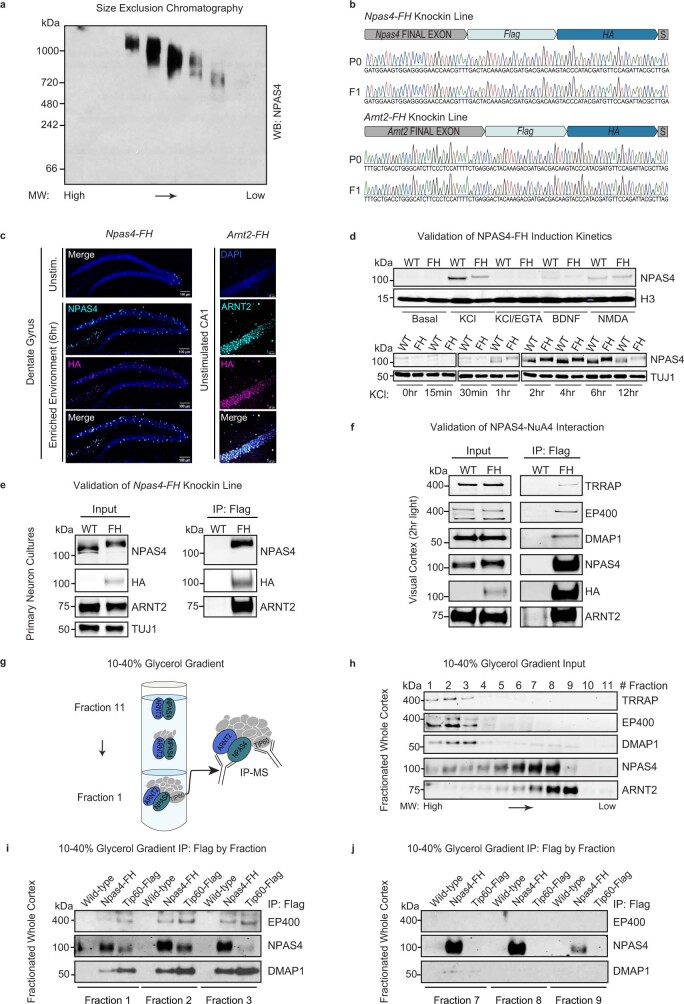
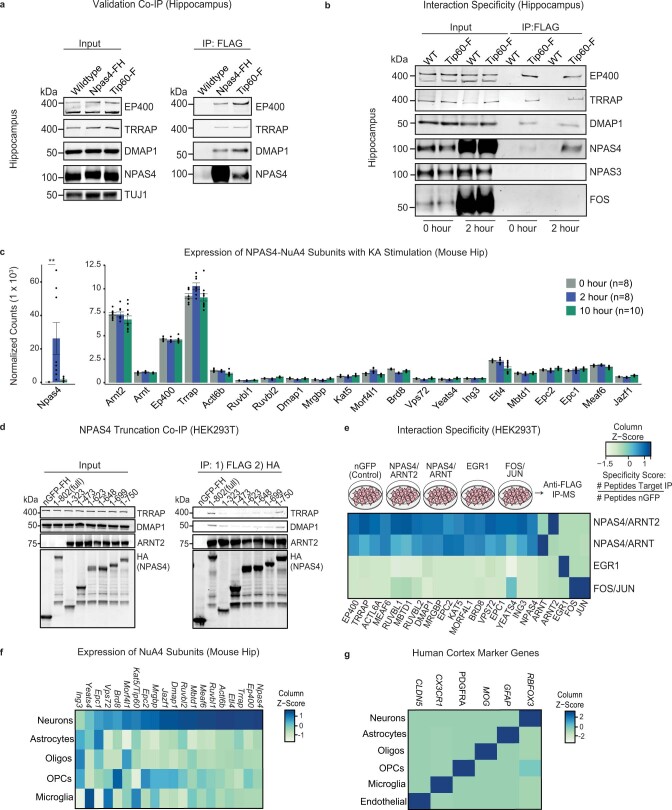
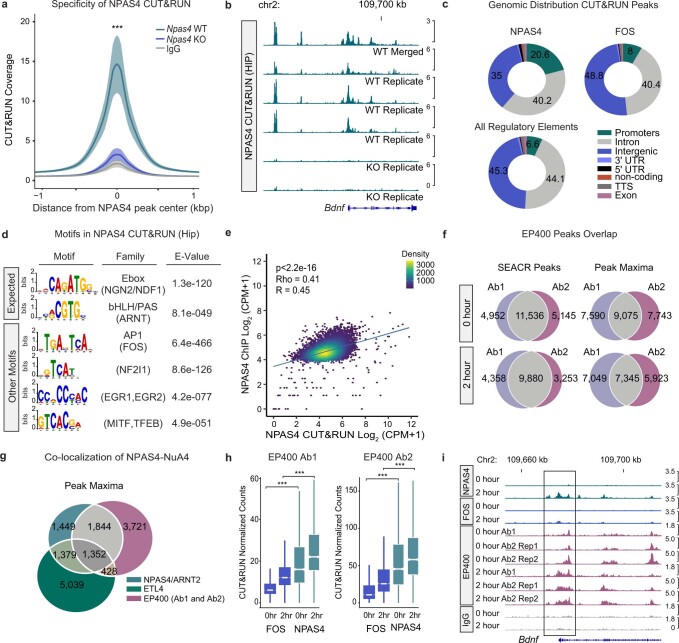
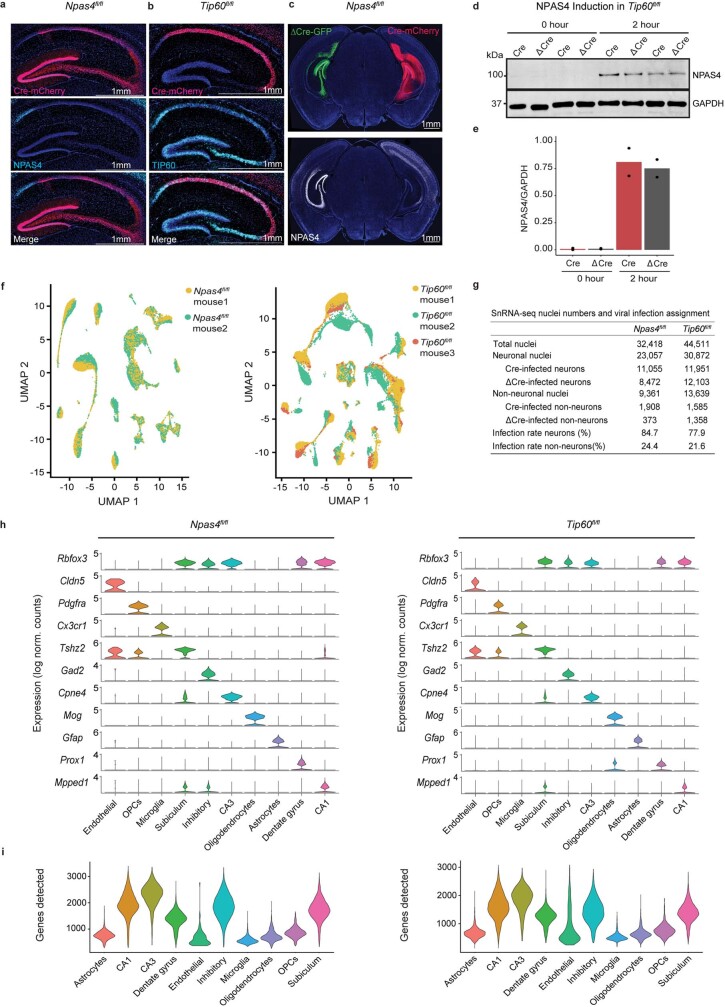
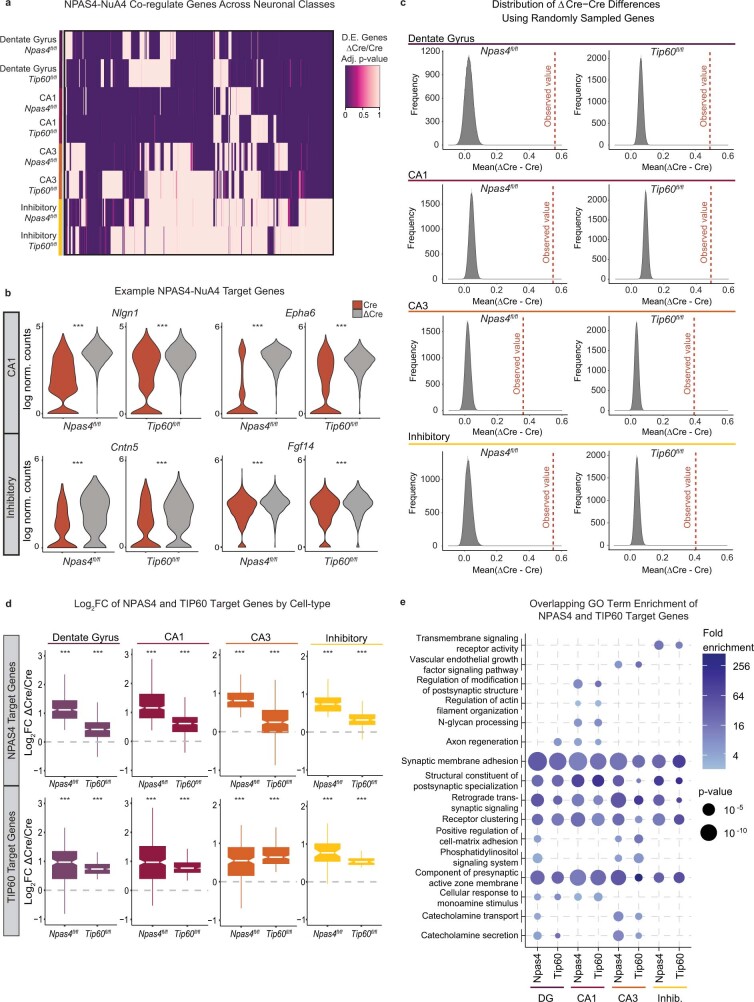
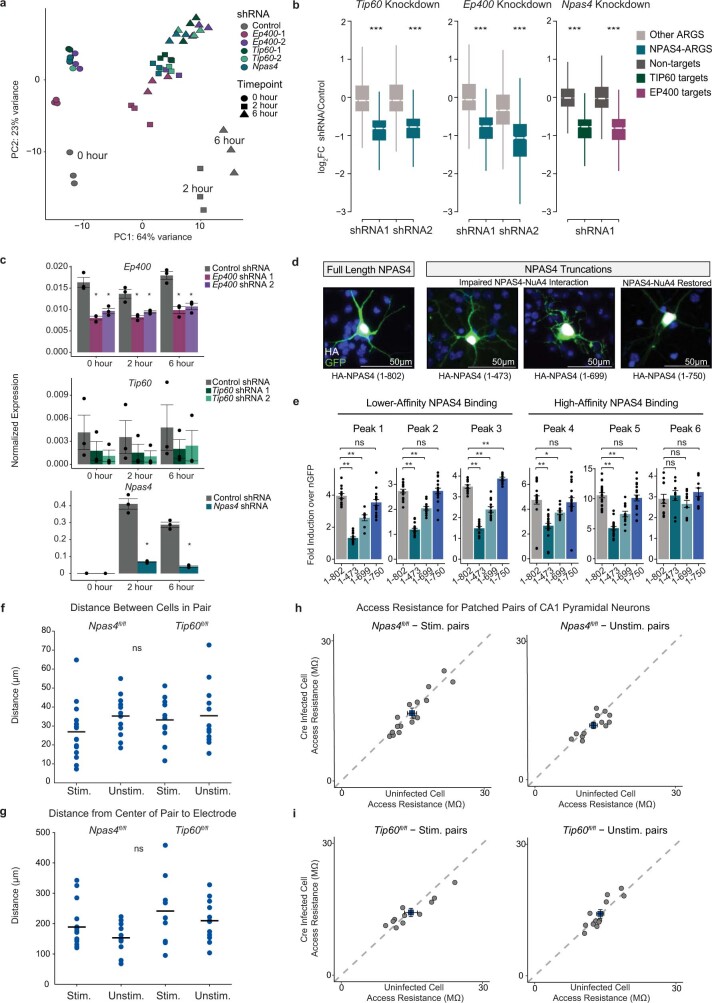
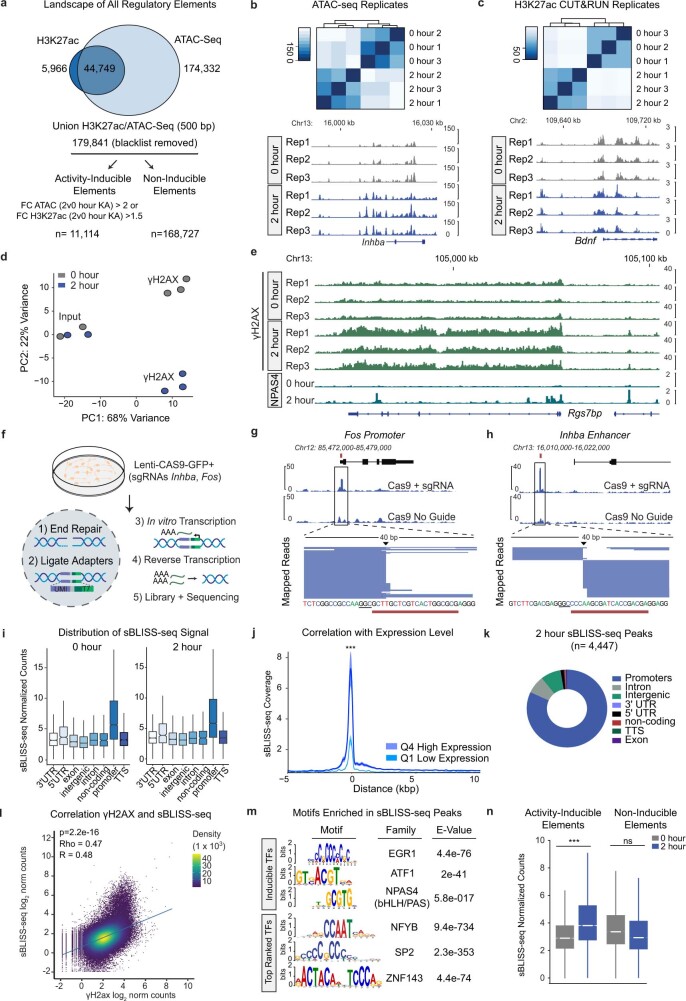
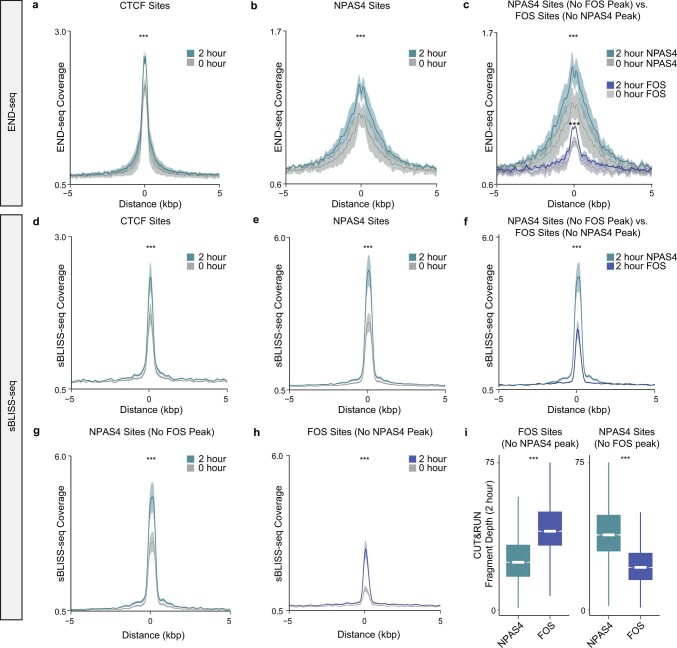
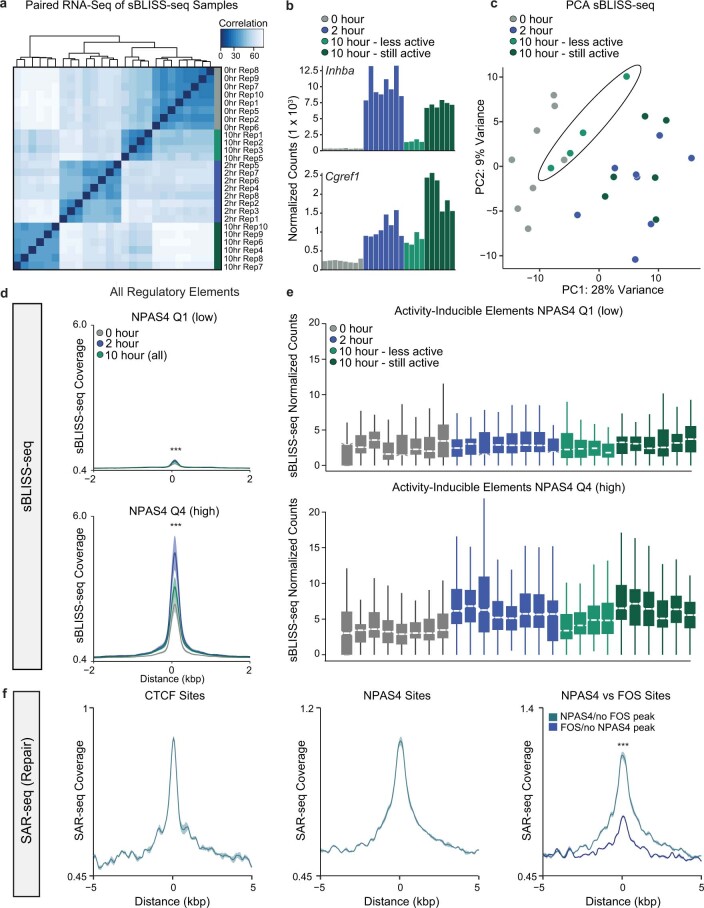
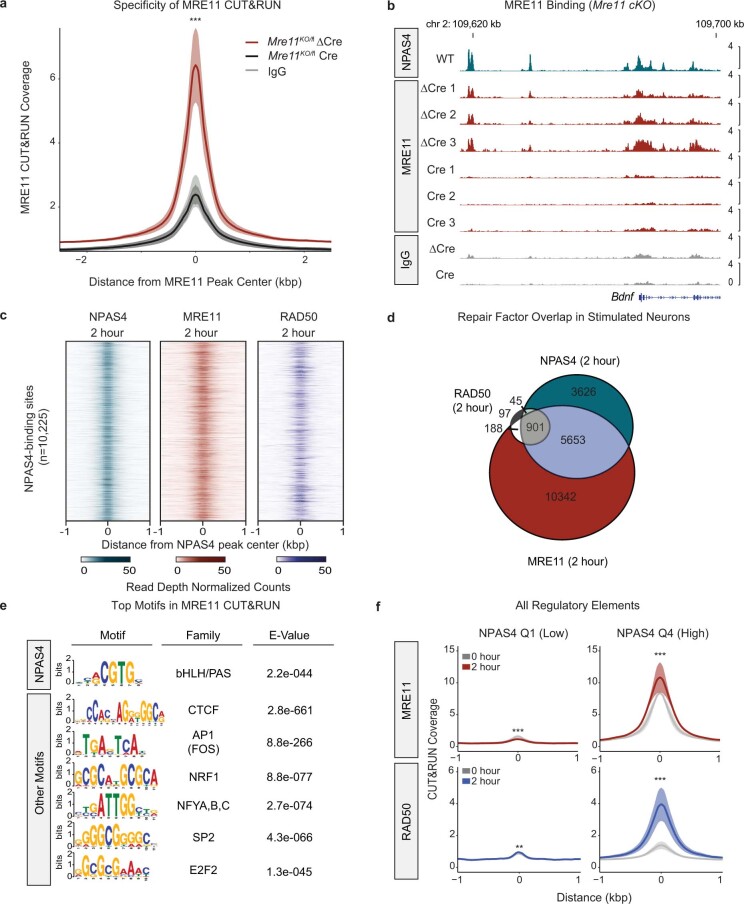
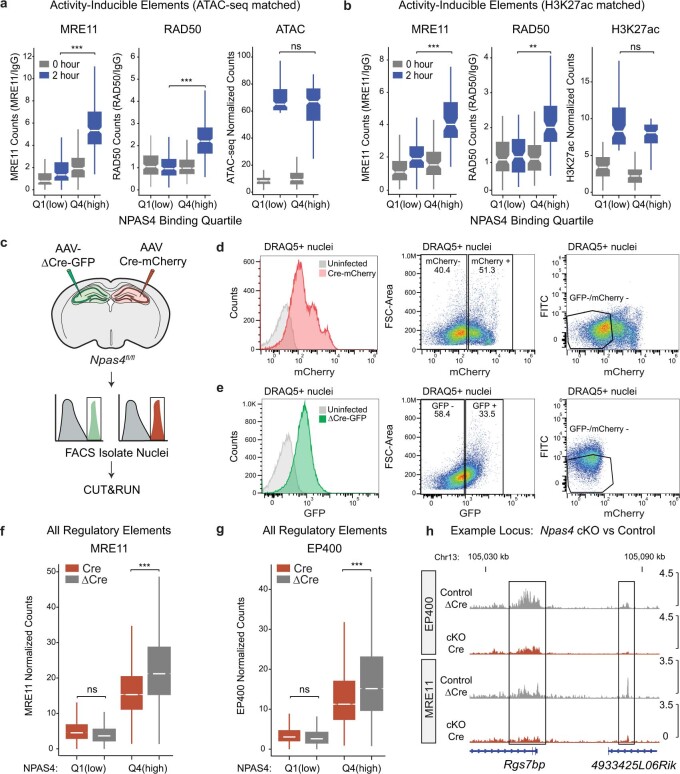
Comment in
-
NPAS4 juggles neuronal activity-dependent transcription and DSB repair with NuA4.Mol Cell. 2023 Apr 20;83(8):1208-1209. doi: 10.1016/j.molcel.2023.03.019. Mol Cell. 2023. PMID: 37084713
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
