

Lysosomal lipid peroxidation regulates tumor immunity
- PMID: 36795483
- PMCID: PMC10104903
- DOI: 10.1172/JCI164596
Lysosomal lipid peroxidation regulates tumor immunity
Abstract
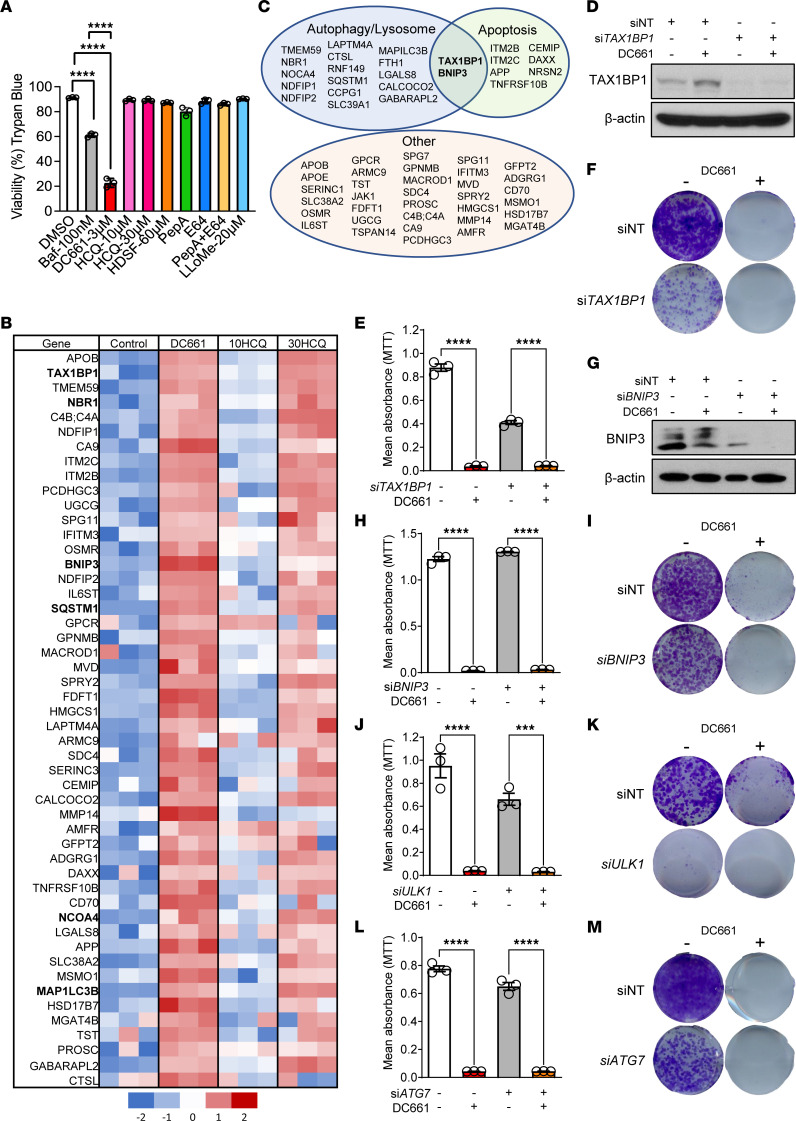
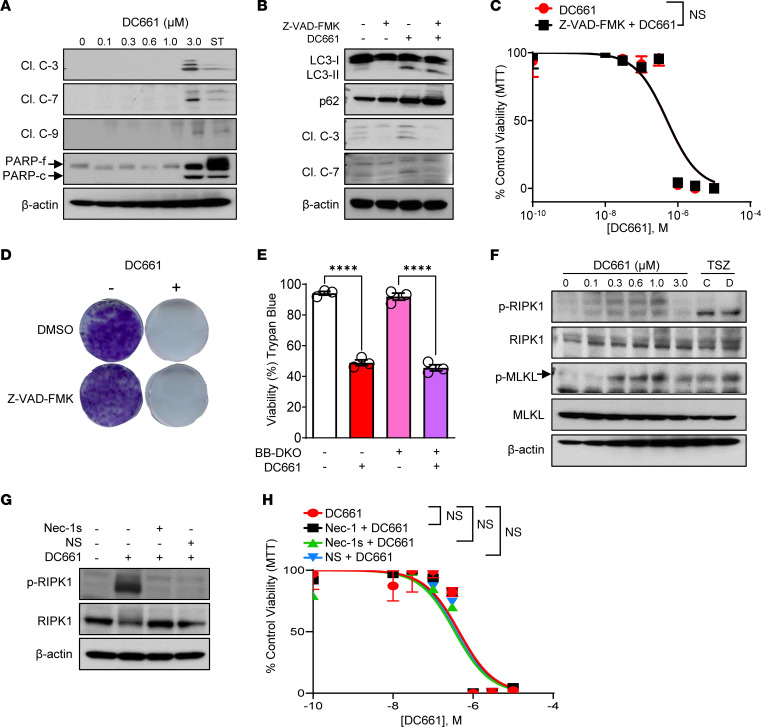
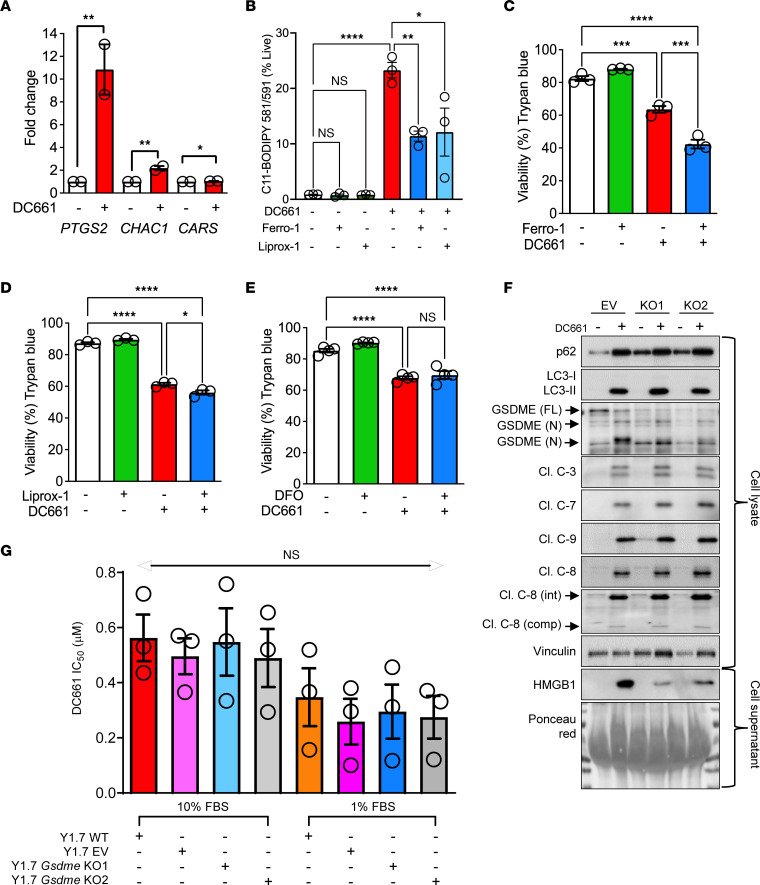
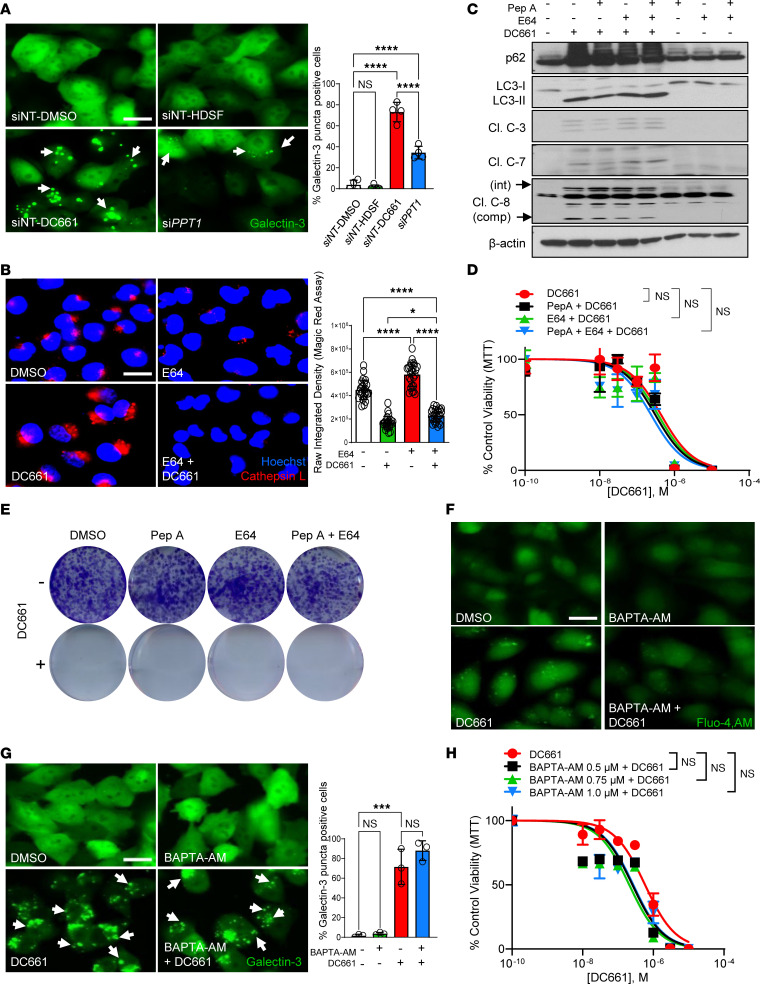
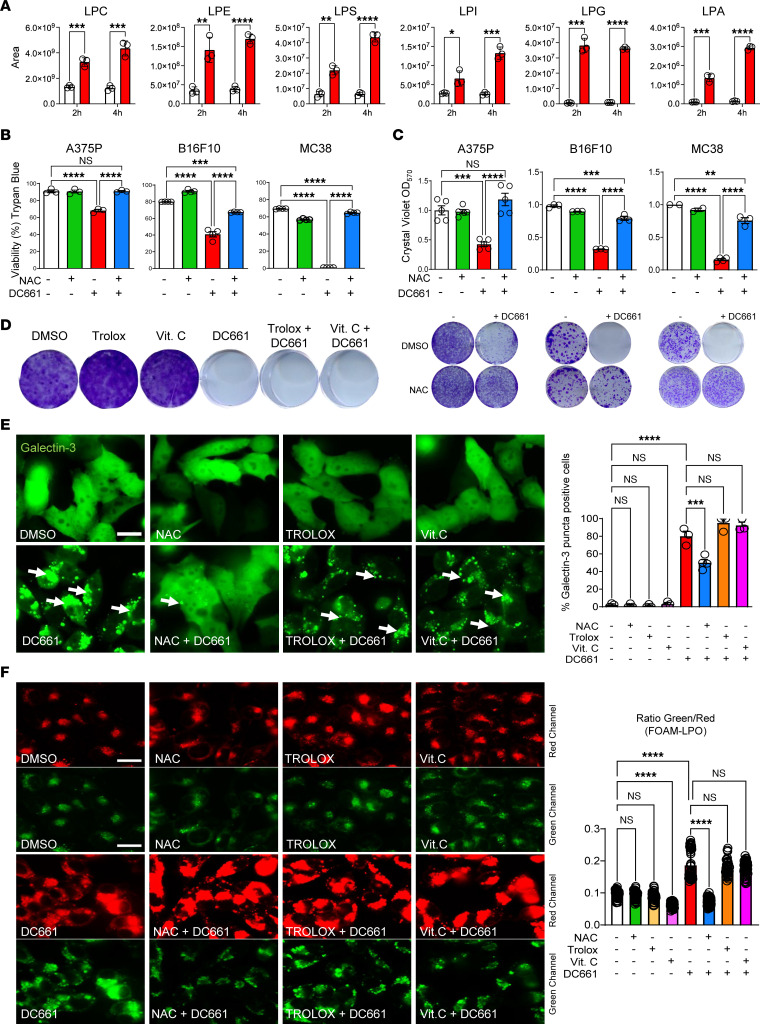
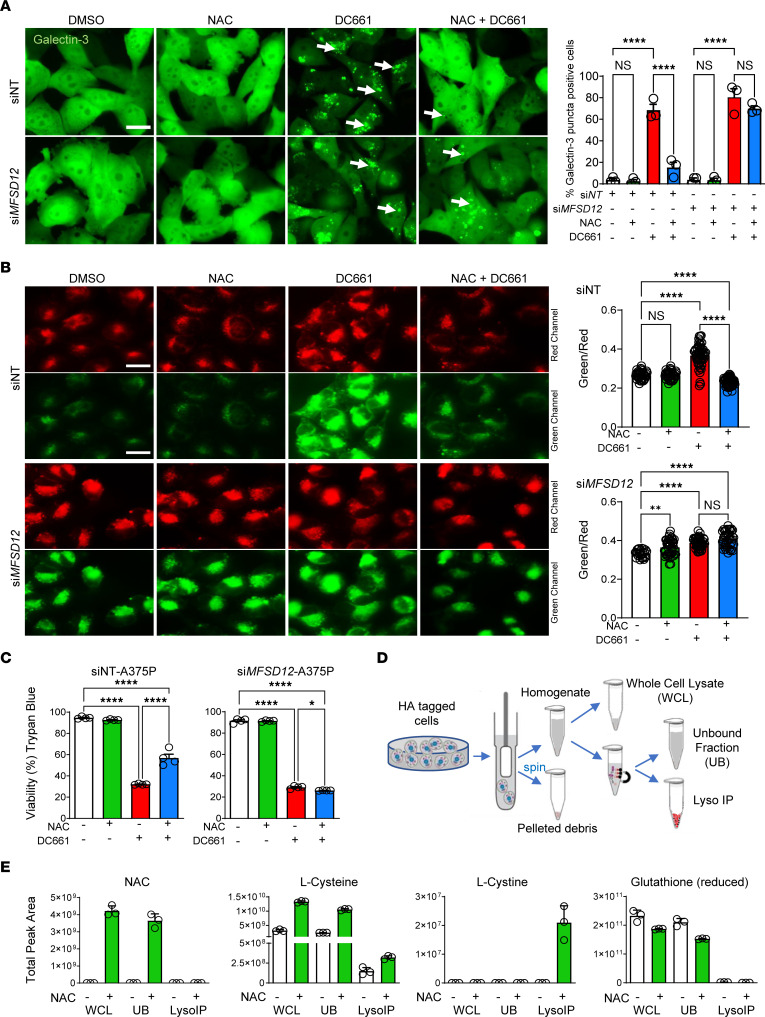
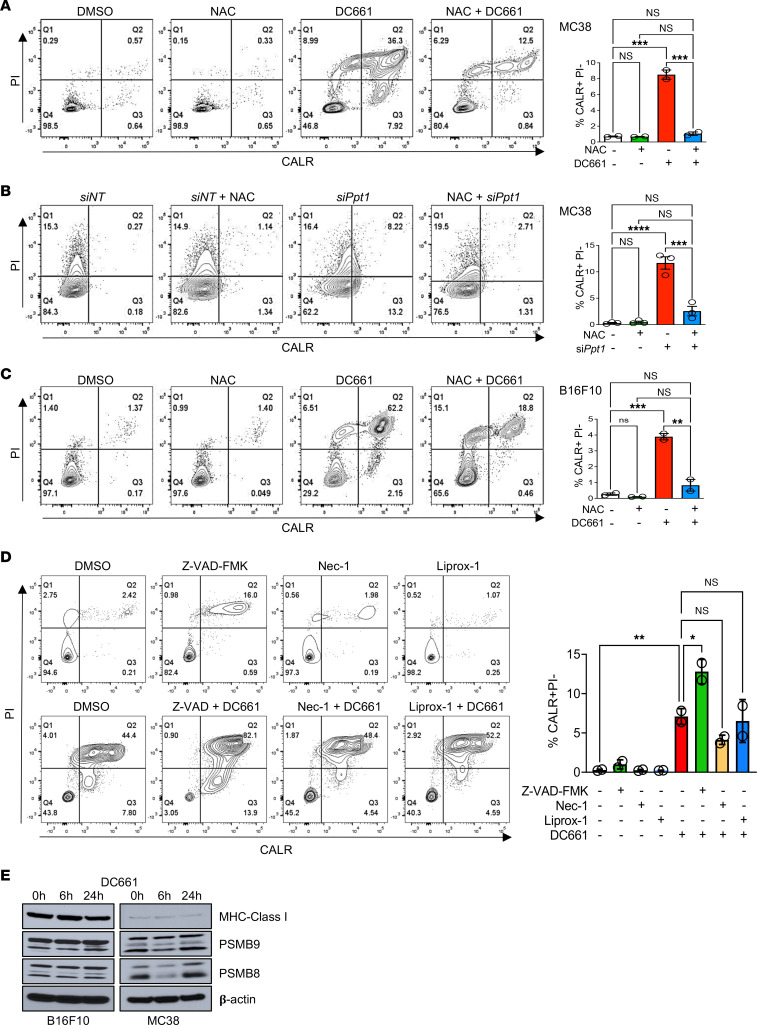
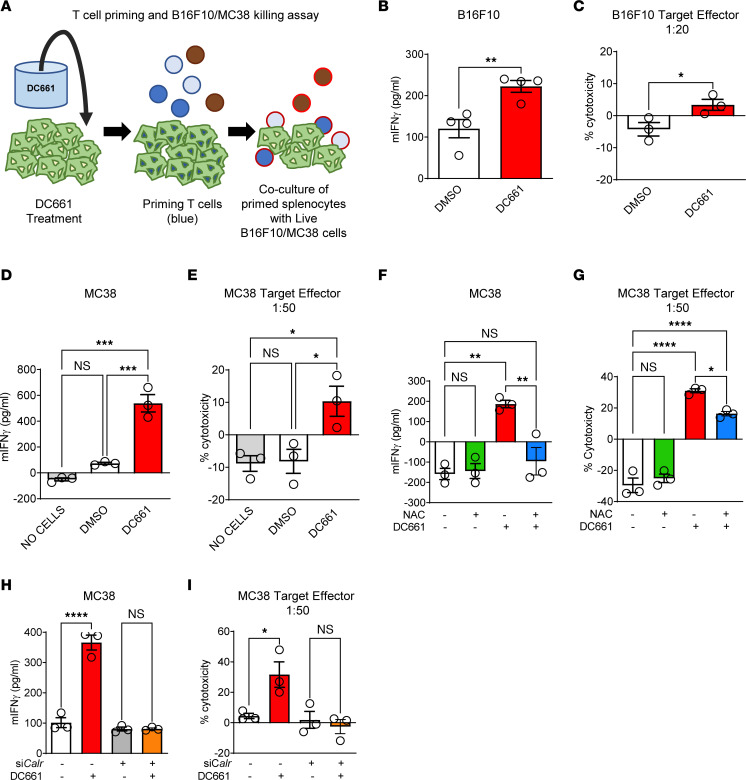
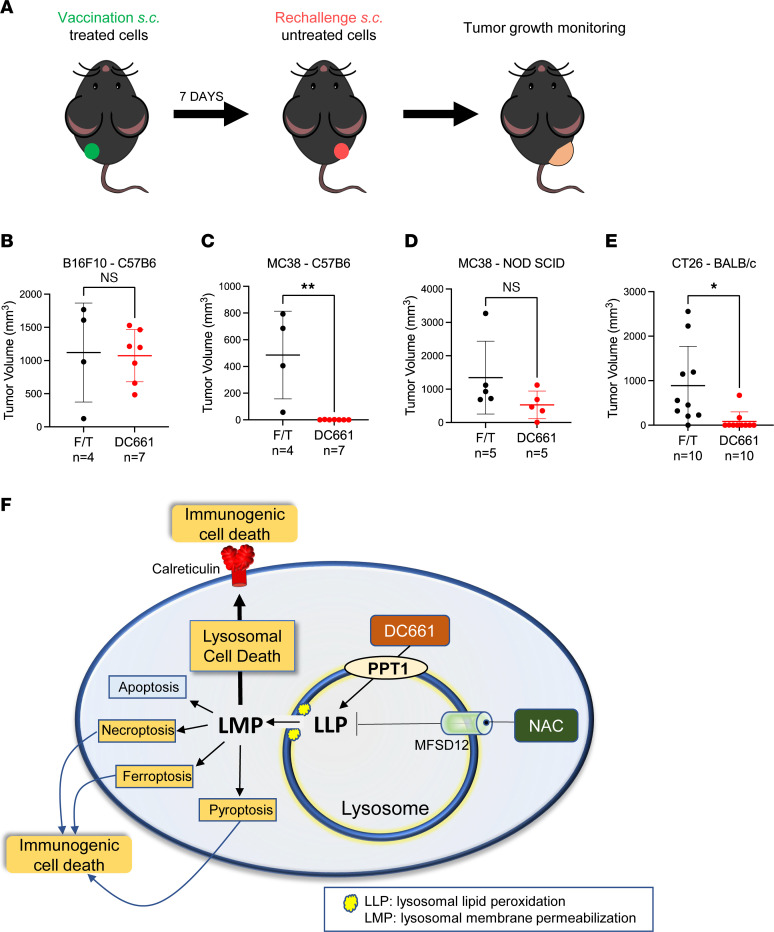
Lysosomal inhibition elicited by palmitoyl-protein thioesterase 1 (PPT1) inhibitors such as DC661 can produce cell death, but the mechanism for this is not completely understood. Programmed cell death pathways (autophagy, apoptosis, necroptosis, ferroptosis, and pyroptosis) were not required to achieve the cytotoxic effect of DC661. Inhibition of cathepsins, or iron or calcium chelation, did not rescue DC661-induced cytotoxicity. PPT1 inhibition induced lysosomal lipid peroxidation (LLP), which led to lysosomal membrane permeabilization and cell death that could be reversed by the antioxidant N-acetylcysteine (NAC) but not by other lipid peroxidation antioxidants. The lysosomal cysteine transporter MFSD12 was required for intralysosomal transport of NAC and rescue of LLP. PPT1 inhibition produced cell-intrinsic immunogenicity with surface expression of calreticulin that could only be reversed with NAC. DC661-treated cells primed naive T cells and enhanced T cell-mediated toxicity. Mice vaccinated with DC661-treated cells engendered adaptive immunity and tumor rejection in "immune hot" tumors but not in "immune cold" tumors. These findings demonstrate that LLP drives lysosomal cell death, a unique immunogenic form of cell death, pointing the way to rational combinations of immunotherapy and lysosomal inhibition that can be tested in clinical trials.
Keywords: Cancer; Cellular immune response; Lysosomes; Oncology.
Figures
Comment in
-
Lysosomal lipid peroxidation mediates immunogenic cell death.J Clin Invest. 2023 Apr 17;133(8):e169240. doi: 10.1172/JCI169240. J Clin Invest. 2023. PMID: 37066873 Free PMC article.
References
-
- Rebecca VW, et al. PPT1 promotes tumor growth and is the molecular target of chloroquine derivatives in cancer. Cancer Discov. 2019;9(2):220–229. doi: 10.1158/2159-8290.CD-18-0706. - DOI - PMC - PubMed
