

On the origin and evolution of RNA editing in metazoans
- PMID: 36795564
- PMCID: PMC9989829
- DOI: 10.1016/j.celrep.2023.112112
On the origin and evolution of RNA editing in metazoans
Abstract
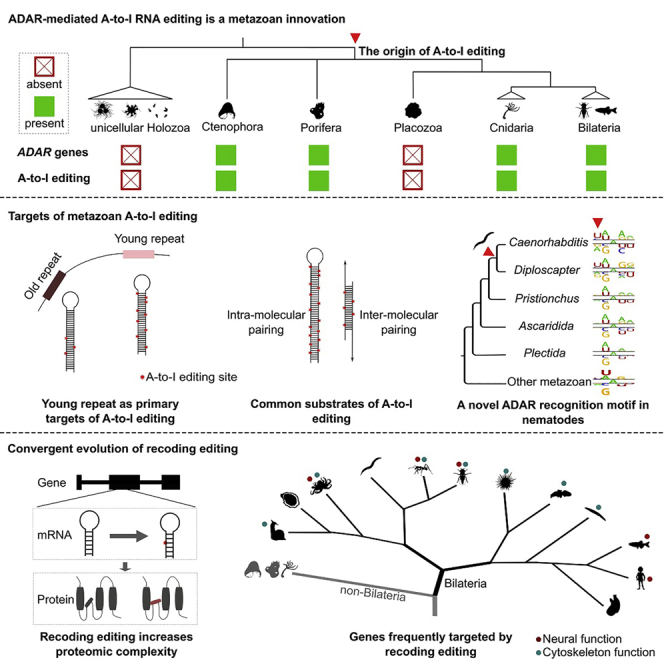
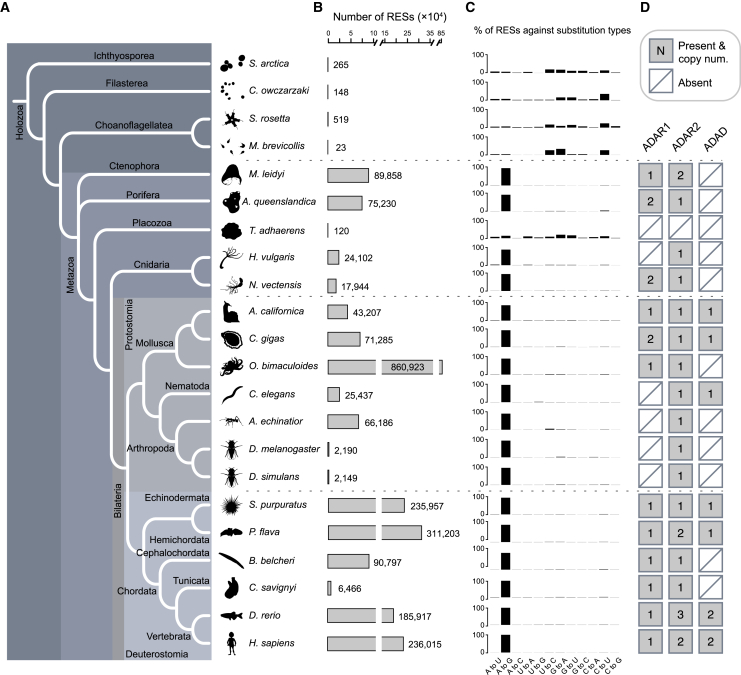
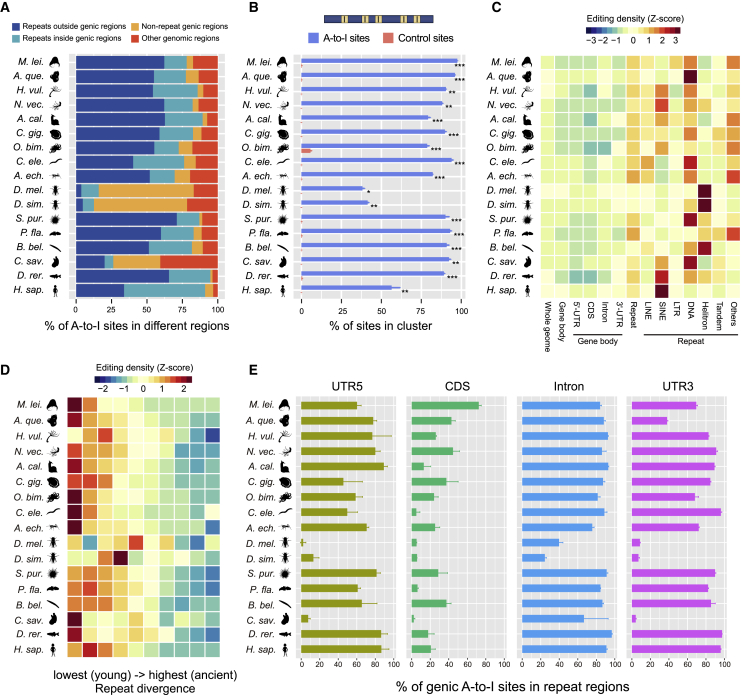
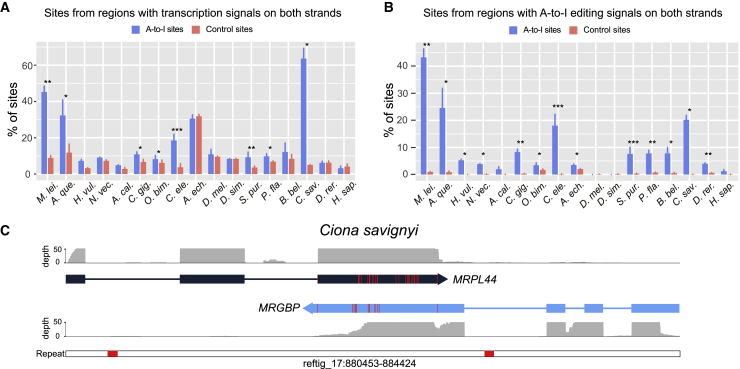
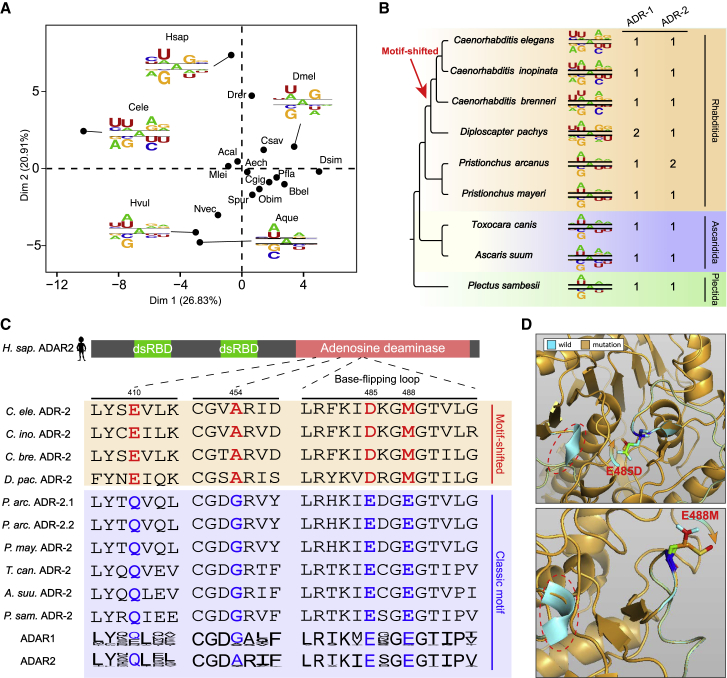
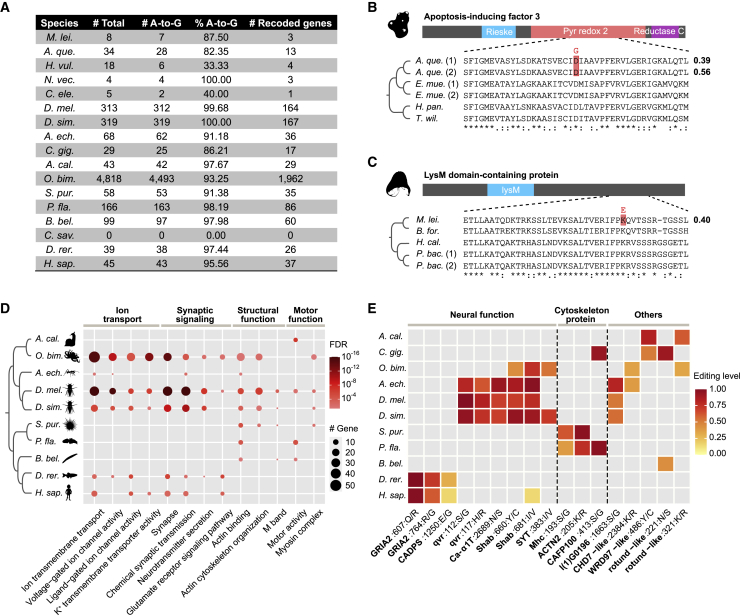
Extensive adenosine-to-inosine (A-to-I) editing of nuclear-transcribed mRNAs is the hallmark of metazoan transcriptional regulation. Here, by profiling the RNA editomes of 22 species that cover major groups of Holozoa, we provide substantial evidence supporting A-to-I mRNA editing as a regulatory innovation originating in the last common ancestor of extant metazoans. This ancient biochemistry process is preserved in most extant metazoan phyla and primarily targets endogenous double-stranded RNA (dsRNA) formed by evolutionarily young repeats. We also find intermolecular pairing of sense-antisense transcripts as an important mechanism for forming dsRNA substrates for A-to-I editing in some but not all lineages. Likewise, recoding editing is rarely shared across lineages but preferentially targets genes involved in neural and cytoskeleton systems in bilaterians. We conclude that metazoan A-to-I editing might first emerge as a safeguard mechanism against repeat-derived dsRNA and was later co-opted into diverse biological processes due to its mutagenic nature.
Keywords: A-to-I editing; Adar; CP: Molecular biology; Holozoa; RNA editing; animal; cytoskeleton; evolution; neural system; recoding editing; sense-antisense.
Copyright © 2023 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
