

Preclinical characterization of pirtobrutinib, a highly selective, noncovalent (reversible) BTK inhibitor
- PMID: 36796019
- PMCID: PMC10651869
- DOI: 10.1182/blood.2022018674
Preclinical characterization of pirtobrutinib, a highly selective, noncovalent (reversible) BTK inhibitor
Abstract
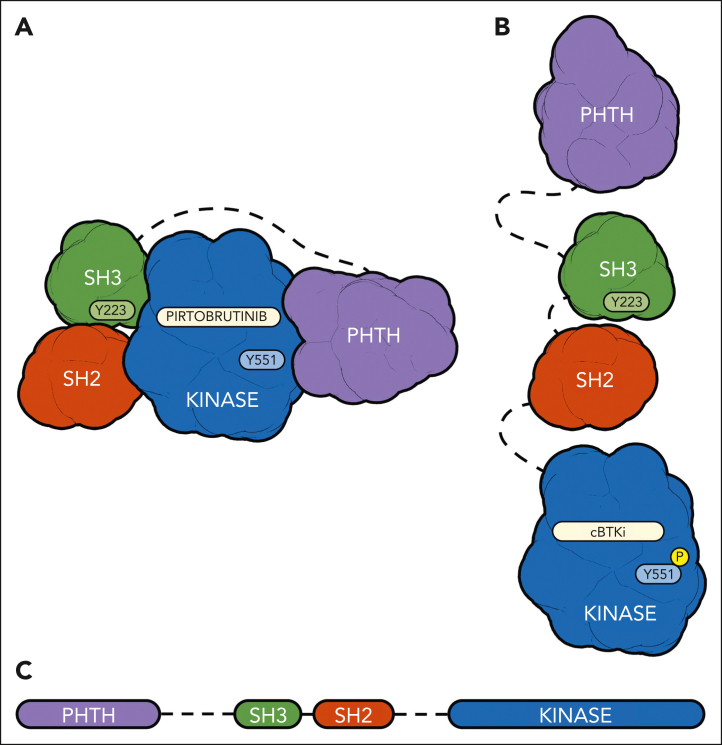
Bruton tyrosine kinase (BTK), a nonreceptor tyrosine kinase, is a major therapeutic target for B-cell-driven malignancies. However, approved covalent BTK inhibitors (cBTKis) are associated with treatment limitations because of off-target side effects, suboptimal oral pharmacology, and development of resistance mutations (eg, C481) that prevent inhibitor binding. Here, we describe the preclinical profile of pirtobrutinib, a potent, highly selective, noncovalent (reversible) BTK inhibitor. Pirtobrutinib binds BTK with an extensive network of interactions to BTK and water molecules in the adenosine triphosphate binding region and shows no direct interaction with C481. Consequently, pirtobrutinib inhibits both BTK and BTK C481 substitution mutants in enzymatic and cell-based assays with similar potencies. In differential scanning fluorimetry studies, BTK bound to pirtobrutinib exhibited a higher melting temperature than cBTKi-bound BTK. Pirtobrutinib, but not cBTKis, prevented Y551 phosphorylation in the activation loop. These data suggest that pirtobrutinib uniquely stabilizes BTK in a closed, inactive conformation. Pirtobrutinib inhibits BTK signaling and cell proliferation in multiple B-cell lymphoma cell lines, and significantly inhibits tumor growth in human lymphoma xenografts in vivo. Enzymatic profiling showed that pirtobrutinib was highly selective for BTK in >98% of the human kinome, and in follow-up cellular studies pirtobrutinib retained >100-fold selectivity over other tested kinases. Collectively, these findings suggest that pirtobrutinib represents a novel BTK inhibitor with improved selectivity and unique pharmacologic, biophysical, and structural attributes with the potential to treat B-cell-driven cancers with improved precision and tolerability. Pirtobrutinib is being tested in phase 3 clinical studies for a variety of B-cell malignancies.
© 2023 by The American Society of Hematology. Licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0), permitting only noncommercial, nonderivative use with attribution. All other rights reserved.
Conflict of interest statement
Conflict-of-interest disclosure: E.B.G., K.E., H.S.R., M.S.R., E.P.C., T.H.M., L.M.H., N.E.B., X.G., J.S., W.W., R.A.W., P.B.A., J.A.B., C.K.A., and B.J.B. are employees of Loxo@Lilly and minor shareholders (restricted stock units) of Eli Lilly and Company. I.L. and K.S.K. are former employees of Loxo@Lilly. K.S.K. reports being a minor shareholder (restricted stock units) of Eli Lilly and Company. B.J.B. reports having patent applications filed related to employment with Loxo@Lilly.
Figures





References
-
- Rawlings DJ, Scharenberg AM, Park H, et al. Activation of BTK by a phosphorylation mechanism initiated by SRC family kinases. Science. 1996;271(5250):822–825. - PubMed
-
- Yang G, Zhou Y, Liu X, et al. A mutation in MYD88 (L265P) supports the survival of lymphoplasmacytic cells by activation of Bruton tyrosine kinase in Waldenstrom macroglobulinemia. Blood. 2013;122(7):1222–1232. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
