

Base editing correction of hypertrophic cardiomyopathy in human cardiomyocytes and humanized mice
- PMID: 36797478
- PMCID: PMC10053064
- DOI: 10.1038/s41591-022-02176-5
Base editing correction of hypertrophic cardiomyopathy in human cardiomyocytes and humanized mice
Abstract
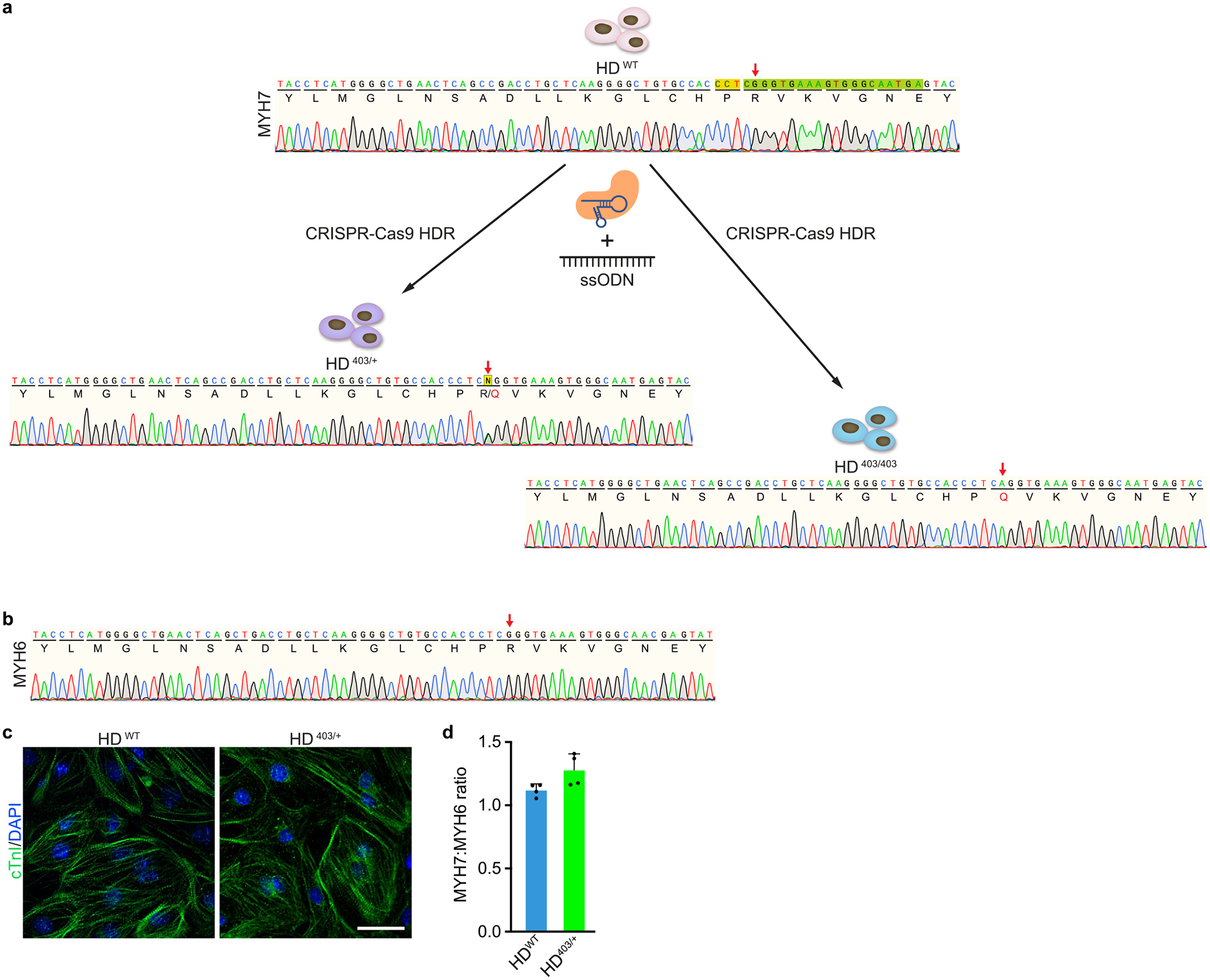
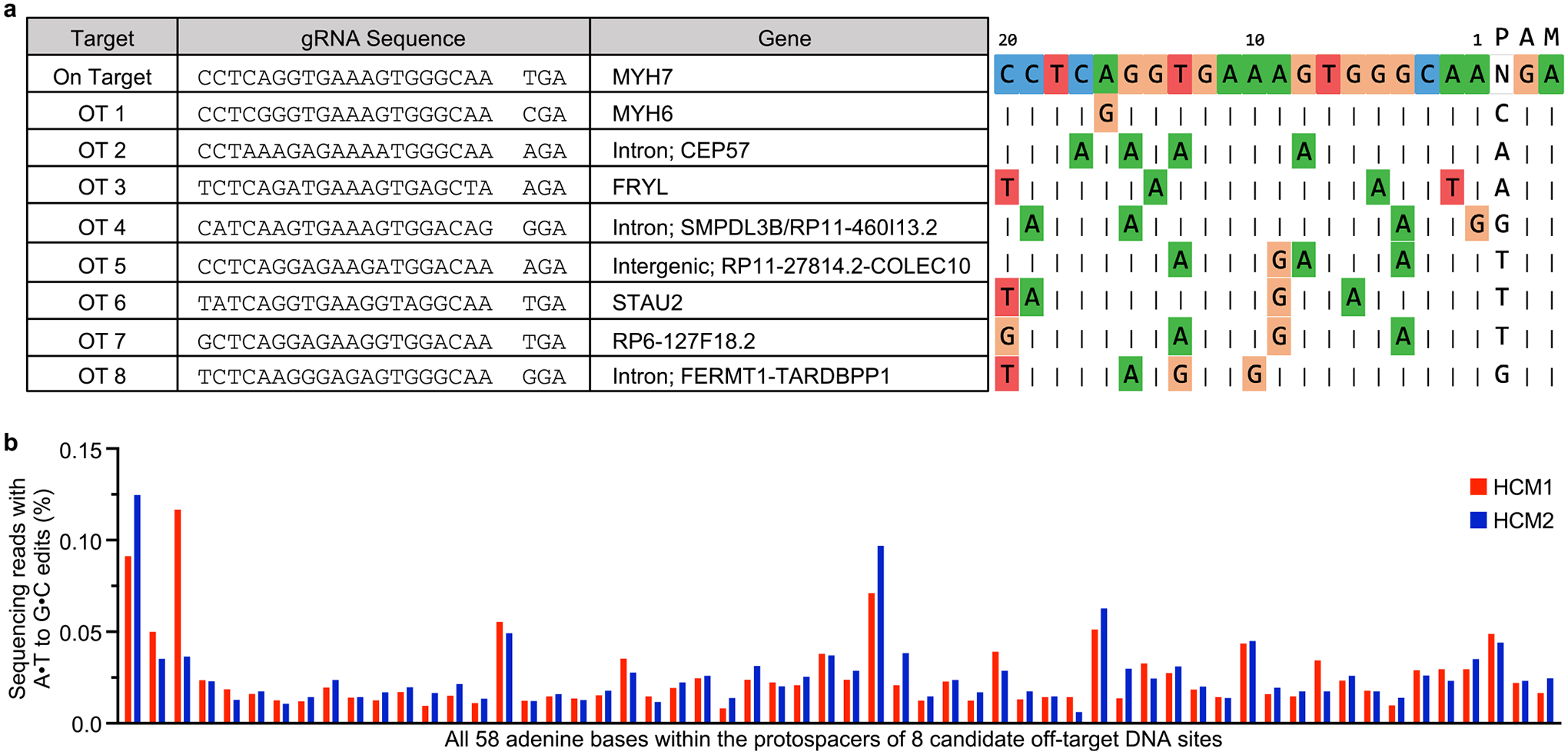
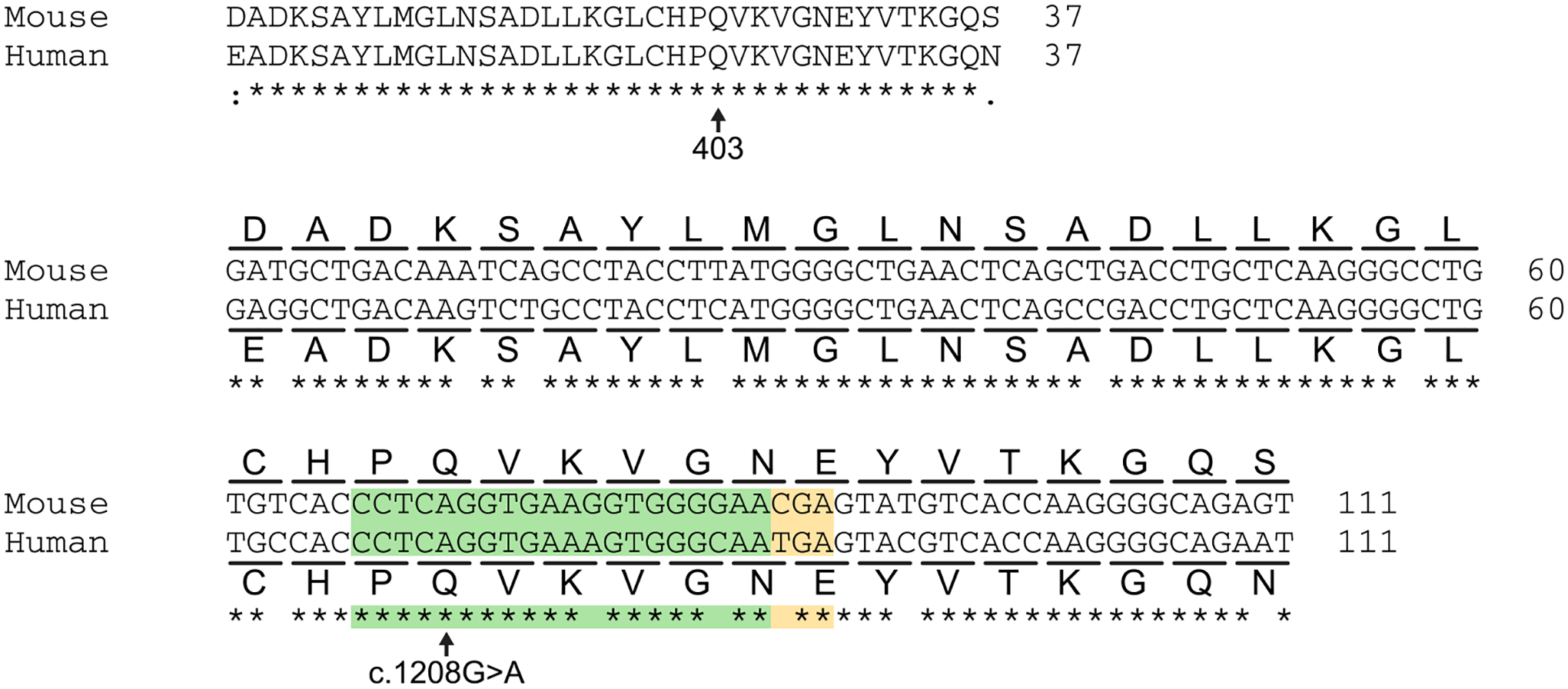
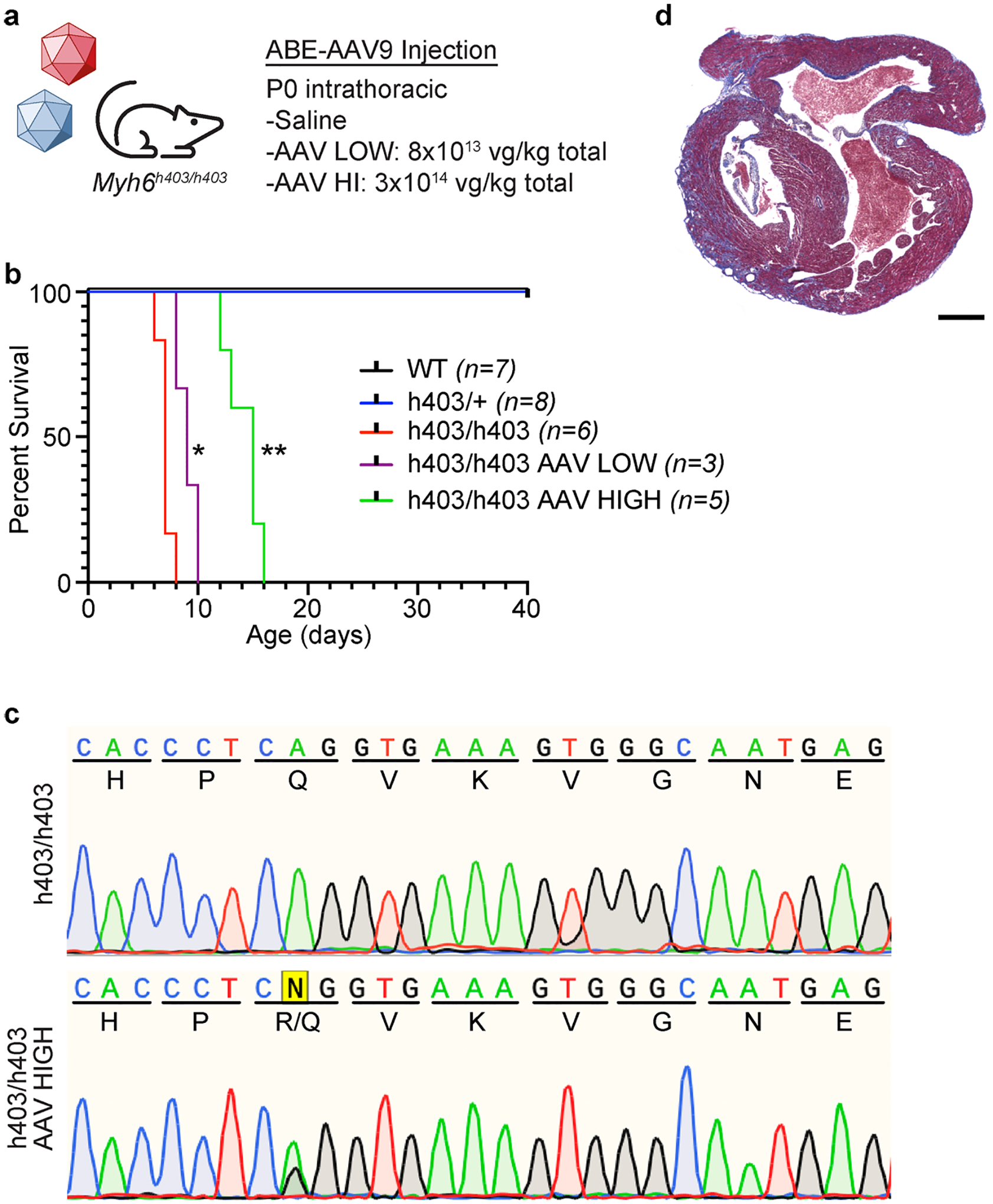
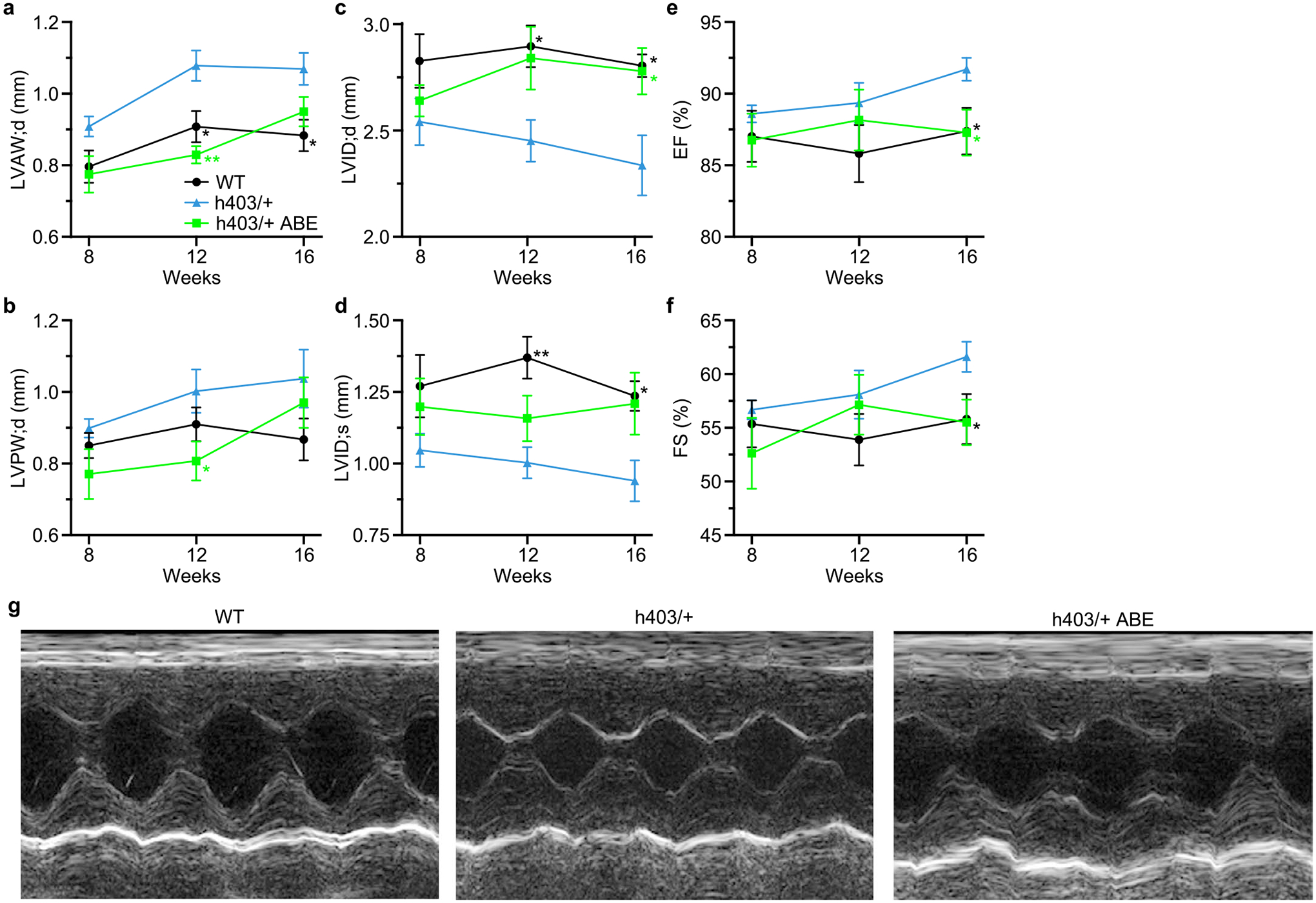
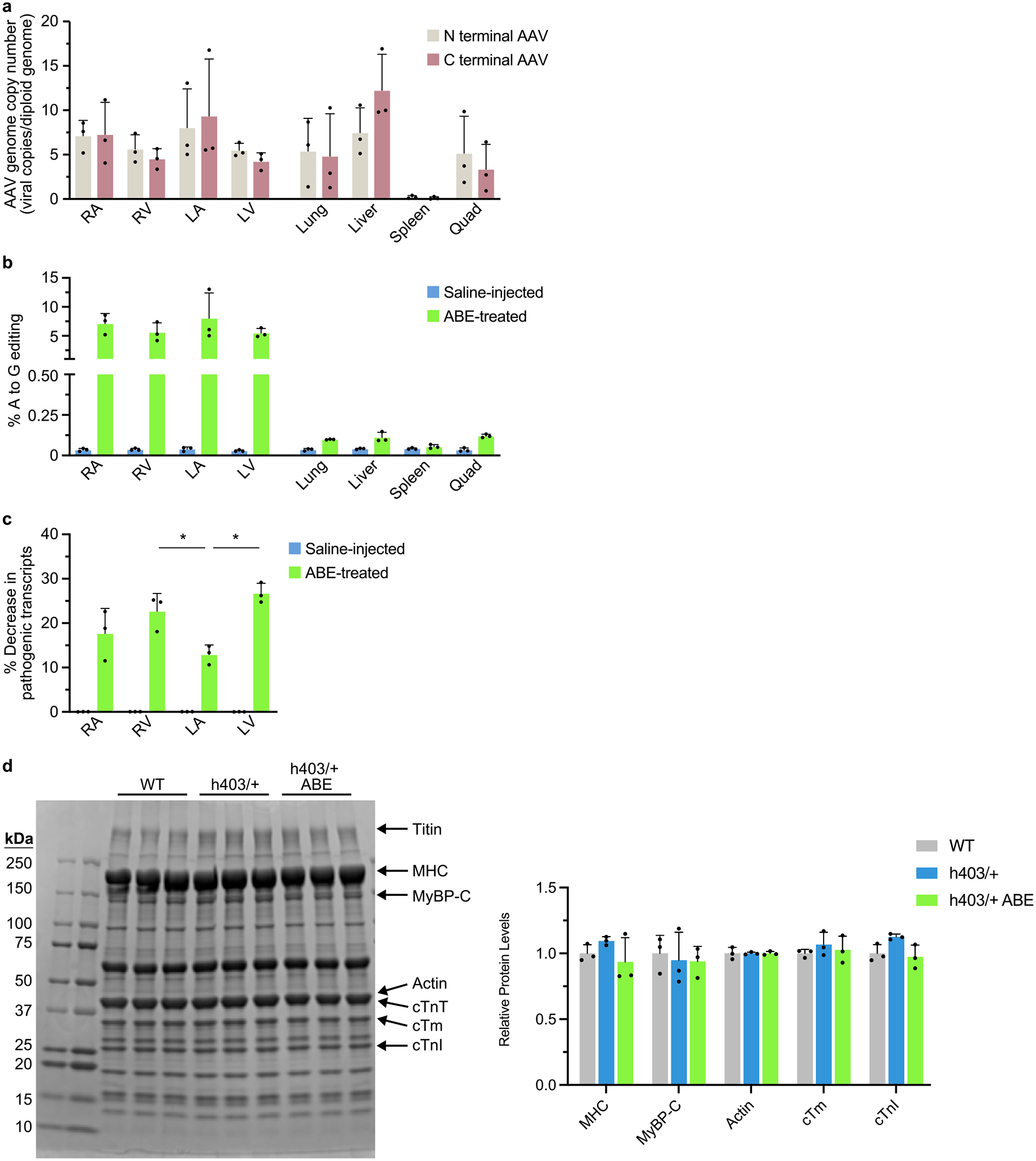
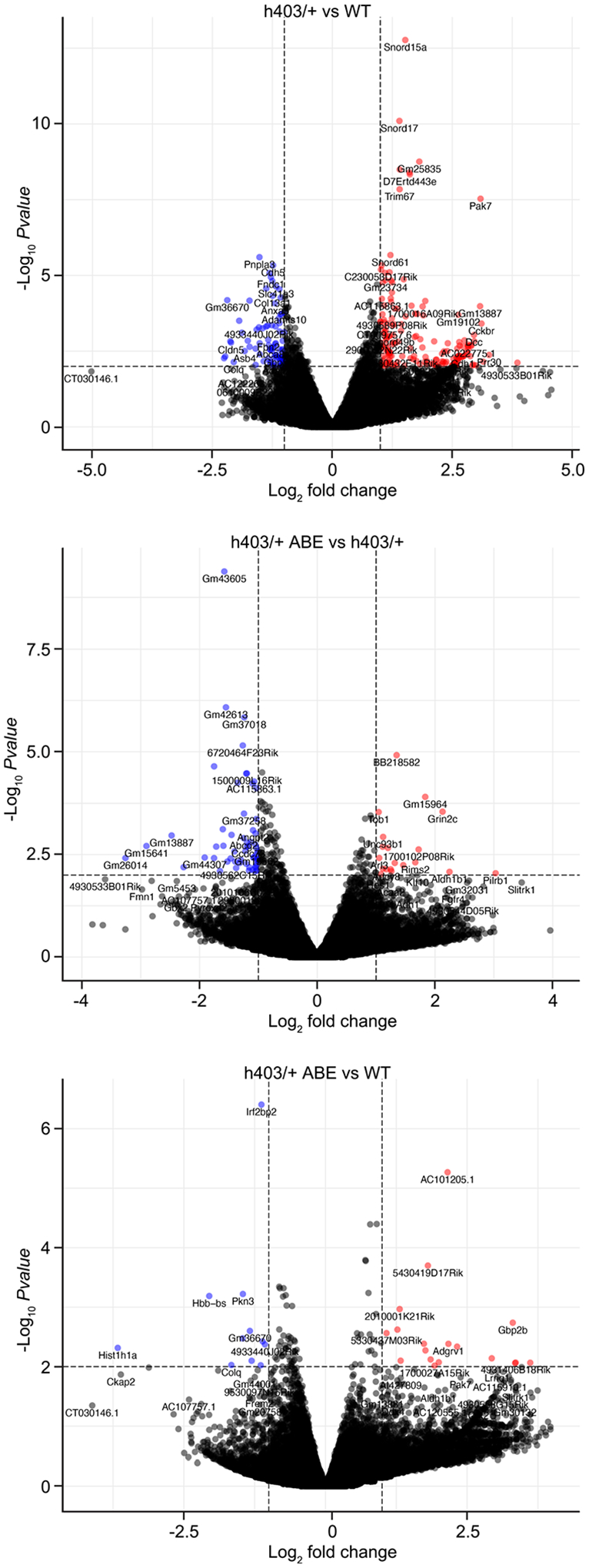
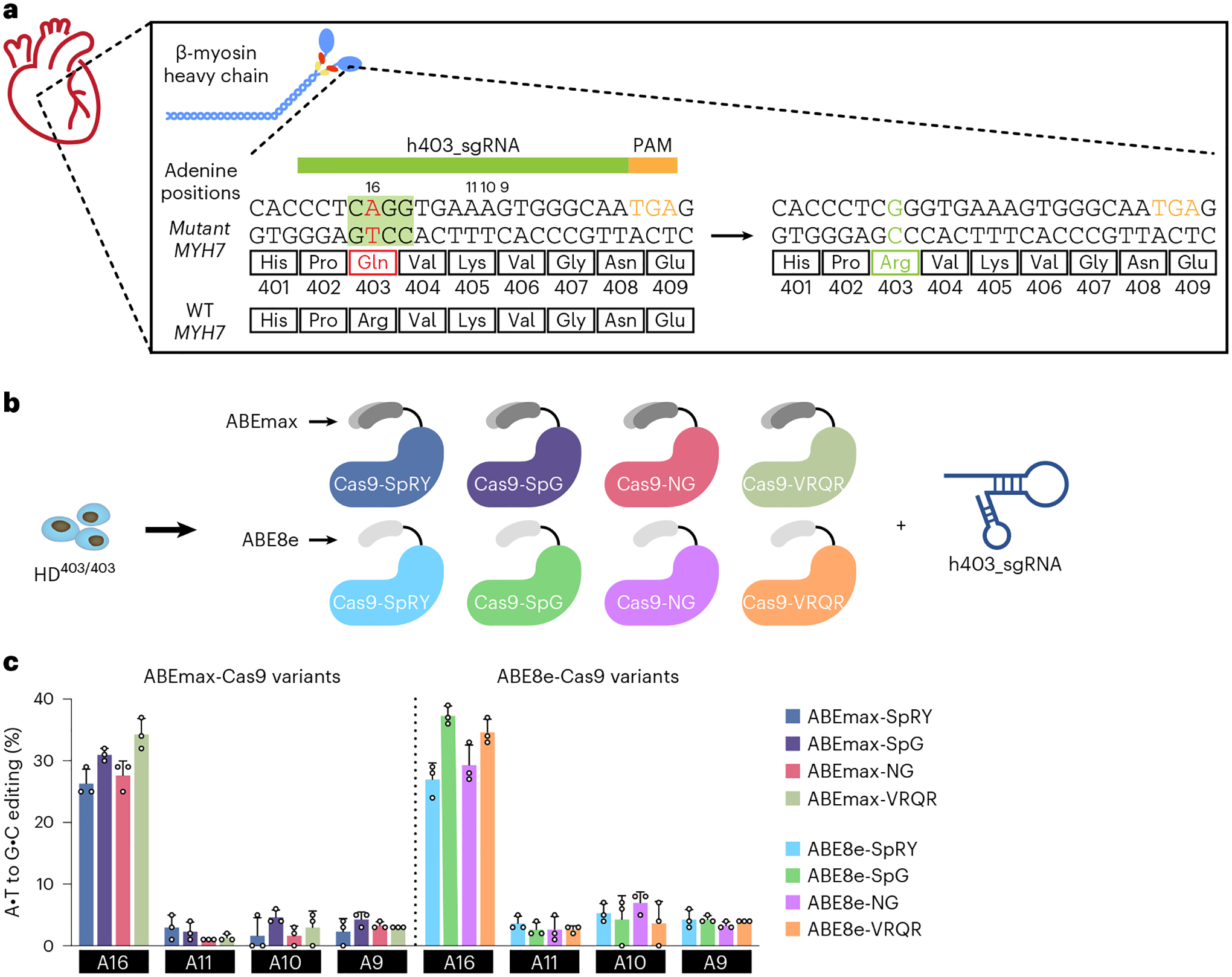
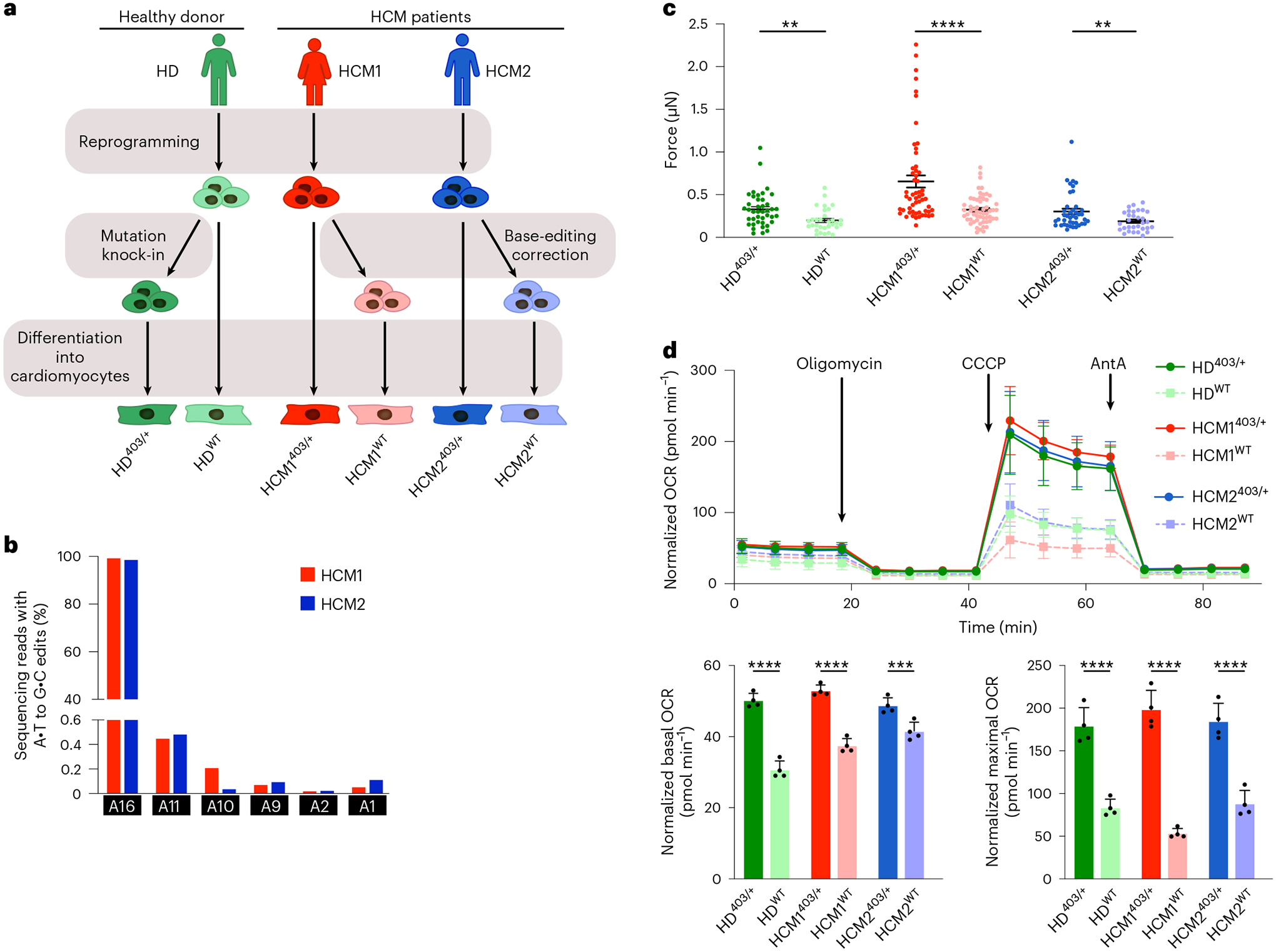
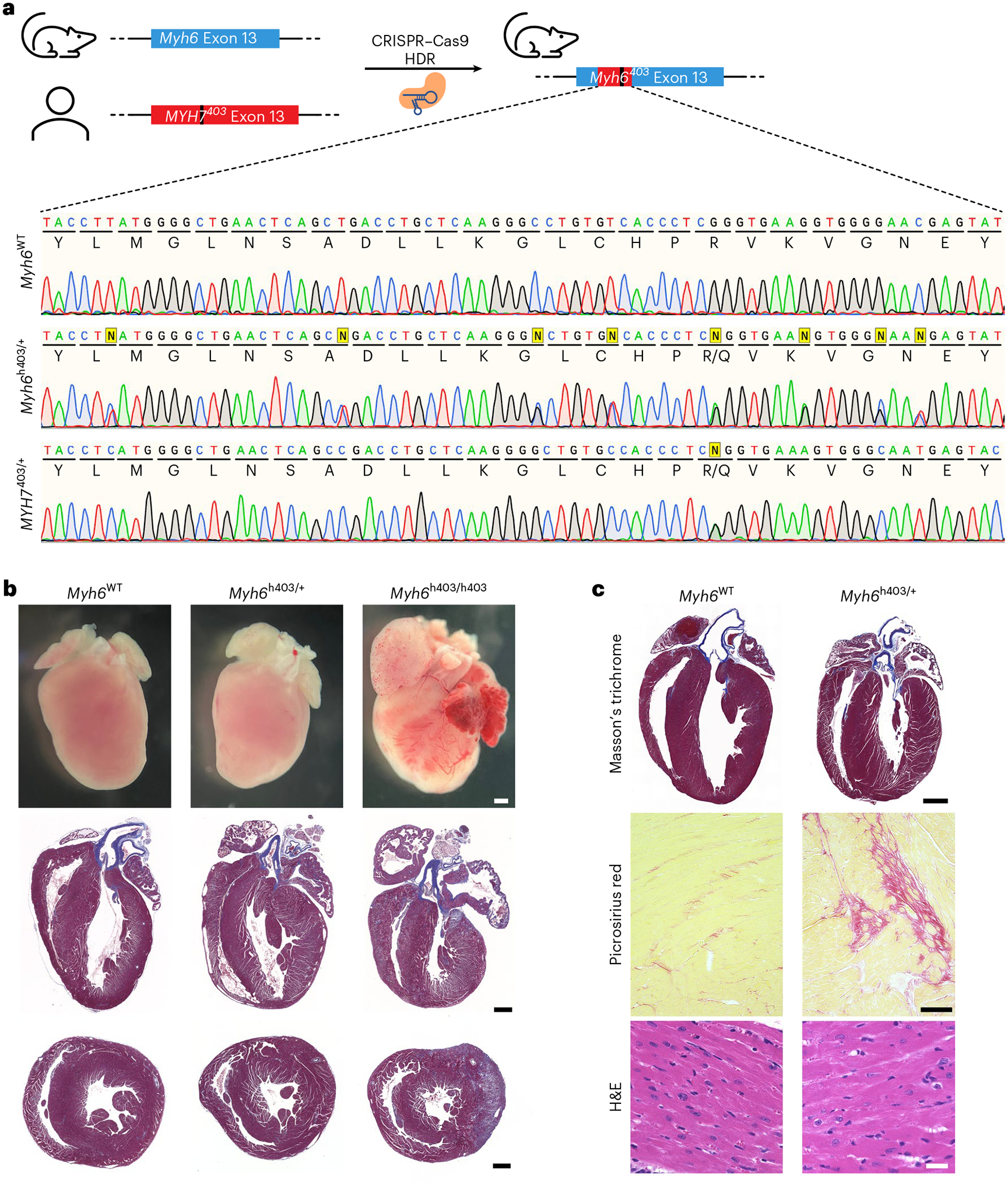
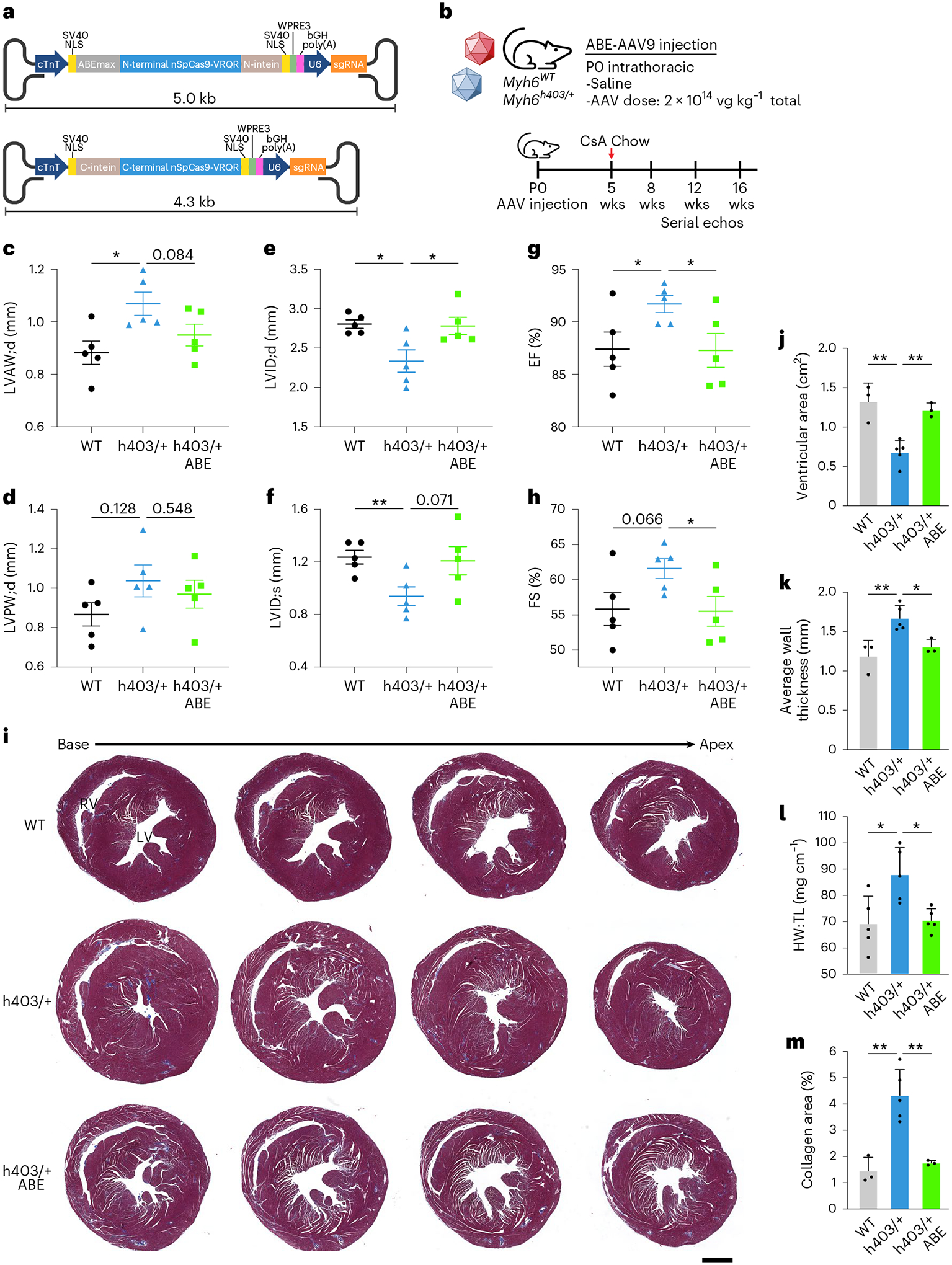
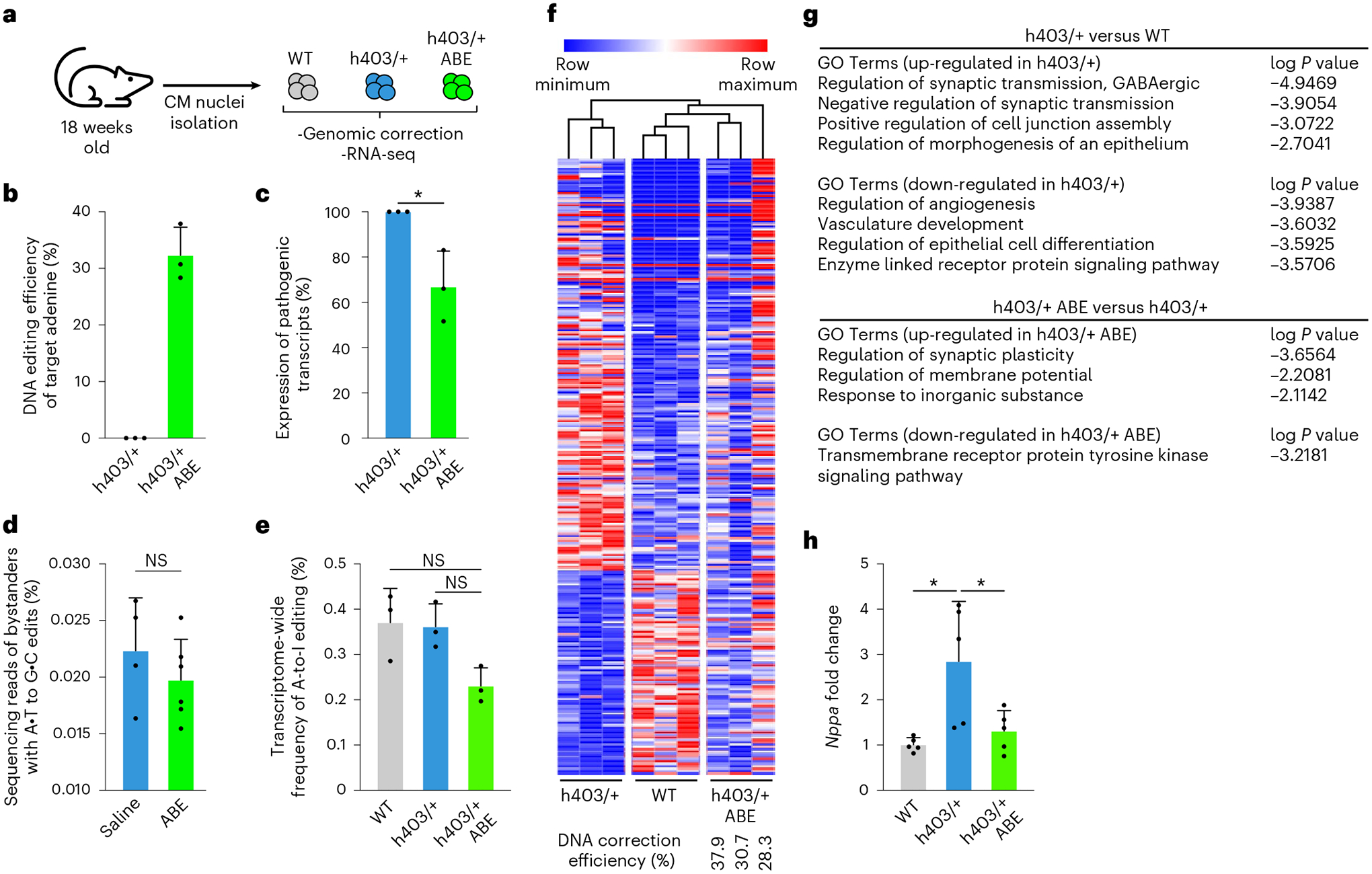
The most common form of genetic heart disease is hypertrophic cardiomyopathy (HCM), which is caused by variants in cardiac sarcomeric genes and leads to abnormal heart muscle thickening. Complications of HCM include heart failure, arrhythmia and sudden cardiac death. The dominant-negative c.1208G>A (p.R403Q) pathogenic variant (PV) in β-myosin (MYH7) is a common and well-studied PV that leads to increased cardiac contractility and HCM onset. In this study we identify an adenine base editor and single-guide RNA system that can efficiently correct this human PV with minimal bystander editing and off-target editing at selected sites. We show that delivery of base editing components rescues pathological manifestations of HCM in induced pluripotent stem cell cardiomyocytes derived from patients with HCM and in a humanized mouse model of HCM. Our findings demonstrate the potential of base editing to treat inherited cardiac diseases and prompt the further development of adenine base editor-based therapies to correct monogenic variants causing cardiac disease.
© 2023. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests
E.N.O. is a consultant for Vertex Pharmaceuticals and Tenaya Therapeutics. The other authors declare no competing interests.
Figures
Comment in
-
CRISPR gene-editing therapies for hypertrophic cardiomyopathy.Nat Med. 2023 Feb;29(2):305-306. doi: 10.1038/s41591-022-02184-5. Nat Med. 2023. PMID: 36797479 Free PMC article.
-
Genome editor tackles disease that can cause sudden death.Nature. 2023 Feb;614(7949):596. doi: 10.1038/d41586-023-00410-9. Nature. 2023. PMID: 36797520 No abstract available.
-
Genome editing prevents hypertrophic cardiomyopathy in mice.Nat Rev Cardiol. 2023 Apr;20(4):211. doi: 10.1038/s41569-023-00852-8. Nat Rev Cardiol. 2023. PMID: 36849814 No abstract available.
-
Testing the genome-editing toolkit in cardiomyopathy.Nat Rev Drug Discov. 2023 Apr;22(4):270. doi: 10.1038/d41573-023-00046-4. Nat Rev Drug Discov. 2023. PMID: 36899271 No abstract available.
-
CRISPRing the hypertrophic cardiomyopathy: correcting one pathogenic variant at a time.Signal Transduct Target Ther. 2023 Jun 26;8(1):254. doi: 10.1038/s41392-023-01526-0. Signal Transduct Target Ther. 2023. PMID: 37365168 Free PMC article. No abstract available.
References
-
- Maron BJ Clinical course and management of hypertrophic cardiomyopathy. N. Engl. J. Med 379, 655–668 (2018). - PubMed
-
- Semsarian C, Ingles J, Maron MS & Maron BJ New perspectives on the prevalence of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol 65, 1249–1254 (2015). - PubMed
-
- Geisterfer-Lowrance AA et al. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell 62, 999–1006 (1990). - PubMed
-
- Tyska MJ et al. Single-molecule mechanics of R403Q cardiac myosin isolated from the mouse model of familial hypertrophic cardiomyopathy. Circ. Res 86, 737–744 (2000). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
