

Prediction of prime editing insertion efficiencies using sequence features and DNA repair determinants
- PMID: 36797492
- PMCID: PMC10567557
- DOI: 10.1038/s41587-023-01678-y
Prediction of prime editing insertion efficiencies using sequence features and DNA repair determinants
Abstract
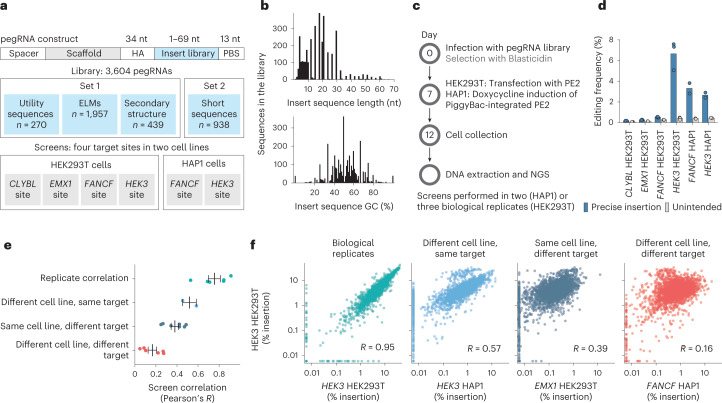
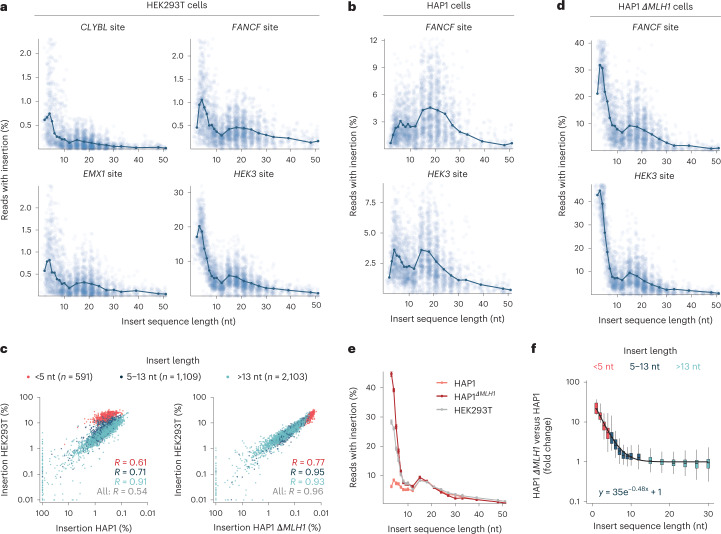
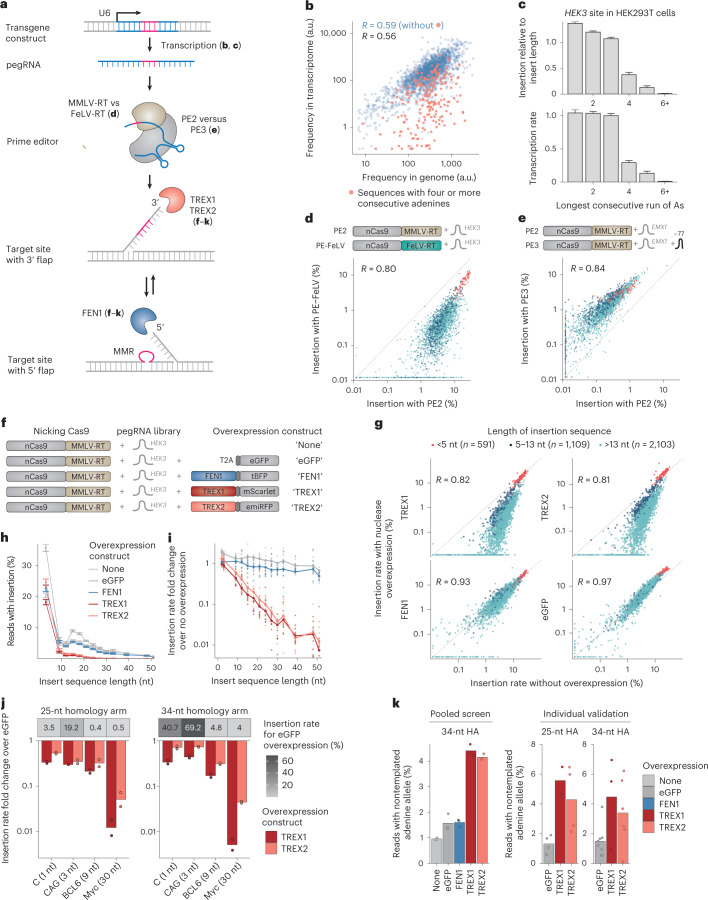
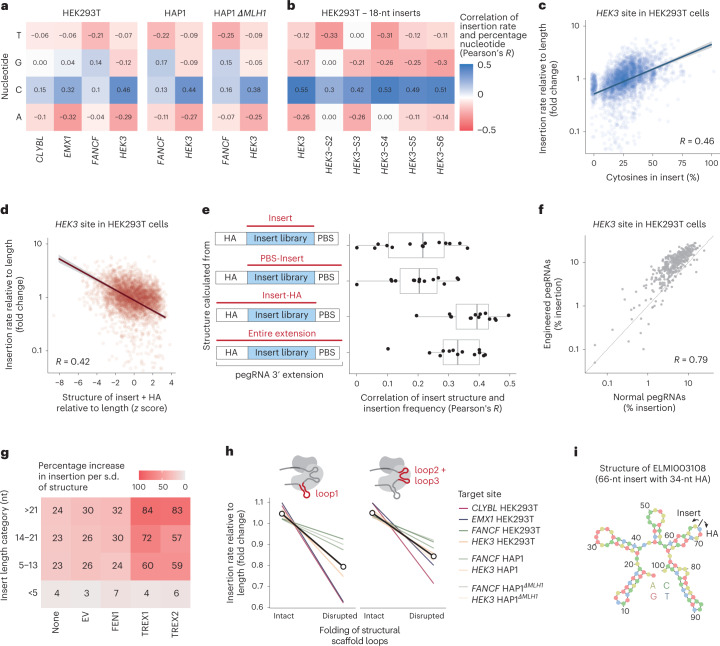
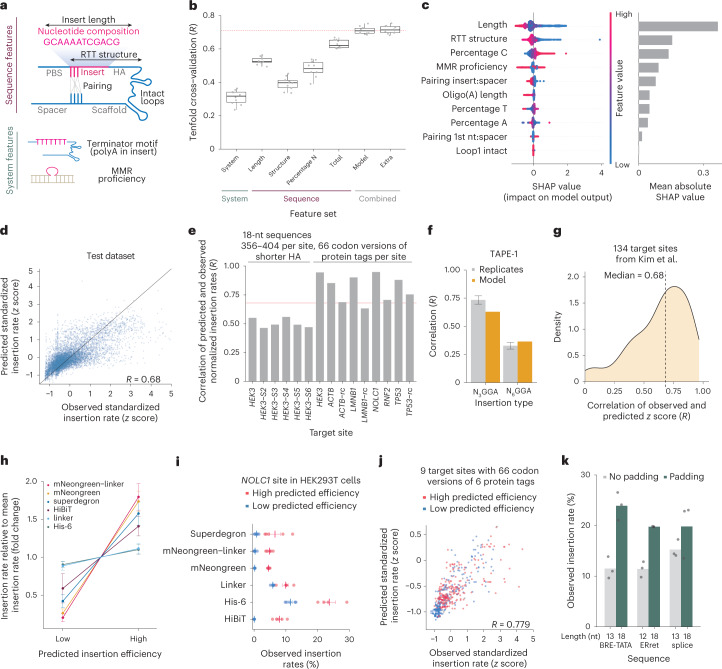
Most short sequences can be precisely written into a selected genomic target using prime editing; however, it remains unclear what factors govern insertion. We design a library of 3,604 sequences of various lengths and measure the frequency of their insertion into four genomic sites in three human cell lines, using different prime editor systems in varying DNA repair contexts. We find that length, nucleotide composition and secondary structure of the insertion sequence all affect insertion rates. We also discover that the 3' flap nucleases TREX1 and TREX2 suppress the insertion of longer sequences. Combining the sequence and repair features into a machine learning model, we can predict relative frequency of insertions into a site with R = 0.70. Finally, we demonstrate how our accurate prediction and user-friendly software help choose codon variants of common fusion tags that insert at high efficiency, and provide a catalog of empirically determined insertion rates for over a hundred useful sequences.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
