

Telomere-to-telomere assembly of diploid chromosomes with Verkko
- PMID: 36797493
- PMCID: PMC10427740
- DOI: 10.1038/s41587-023-01662-6
Telomere-to-telomere assembly of diploid chromosomes with Verkko
Abstract
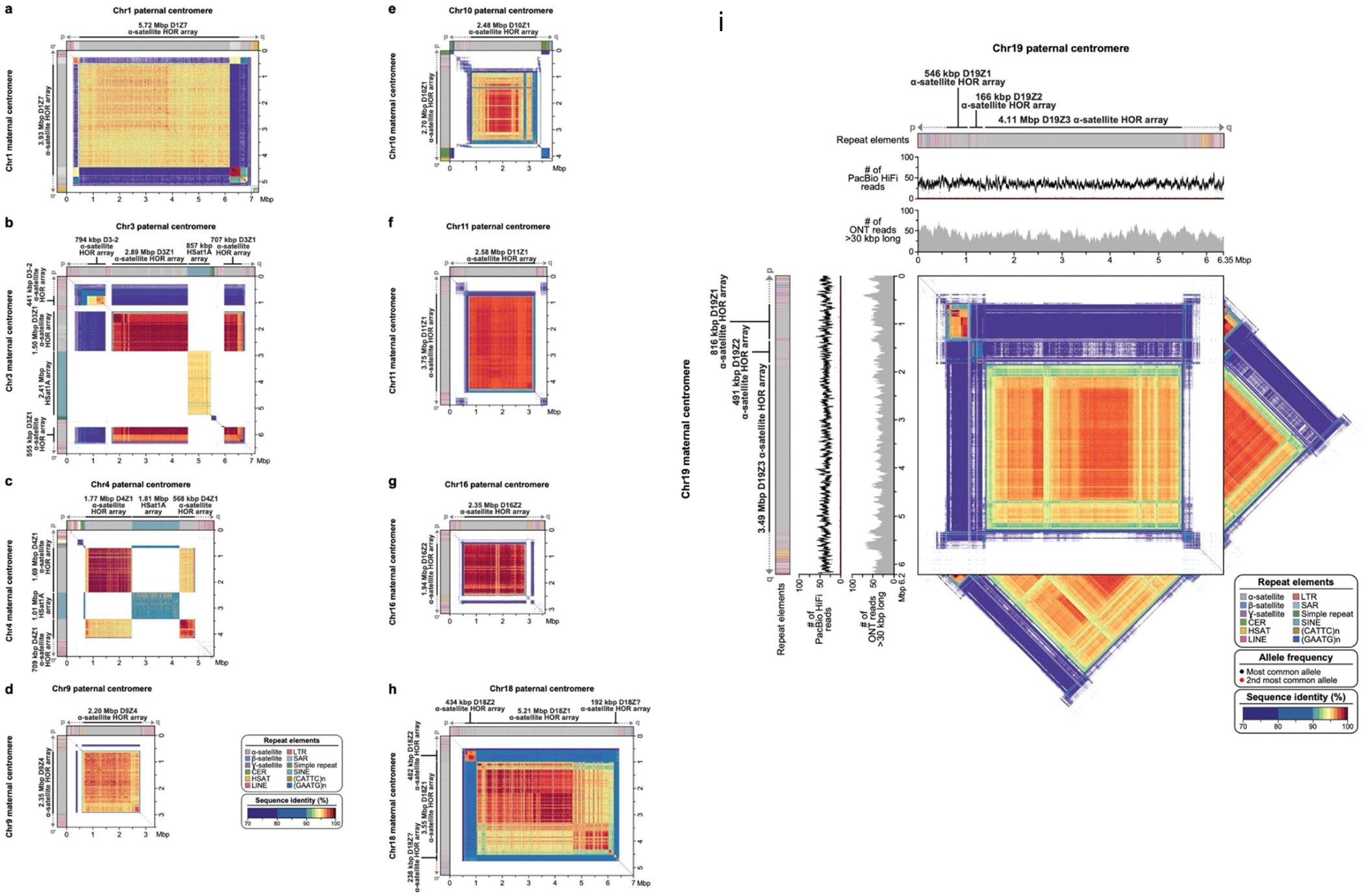
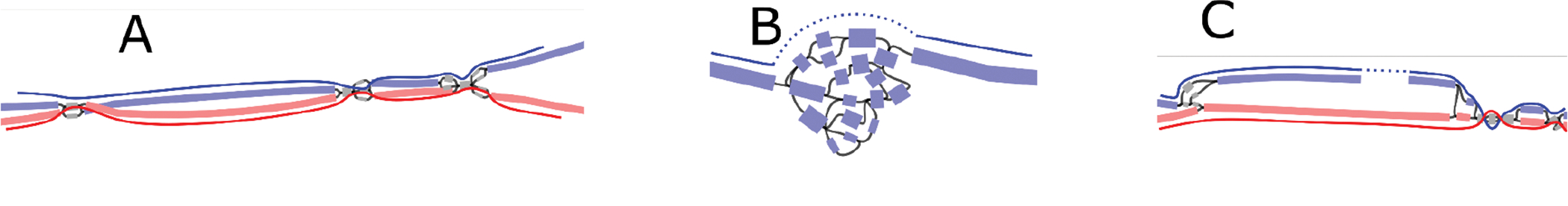
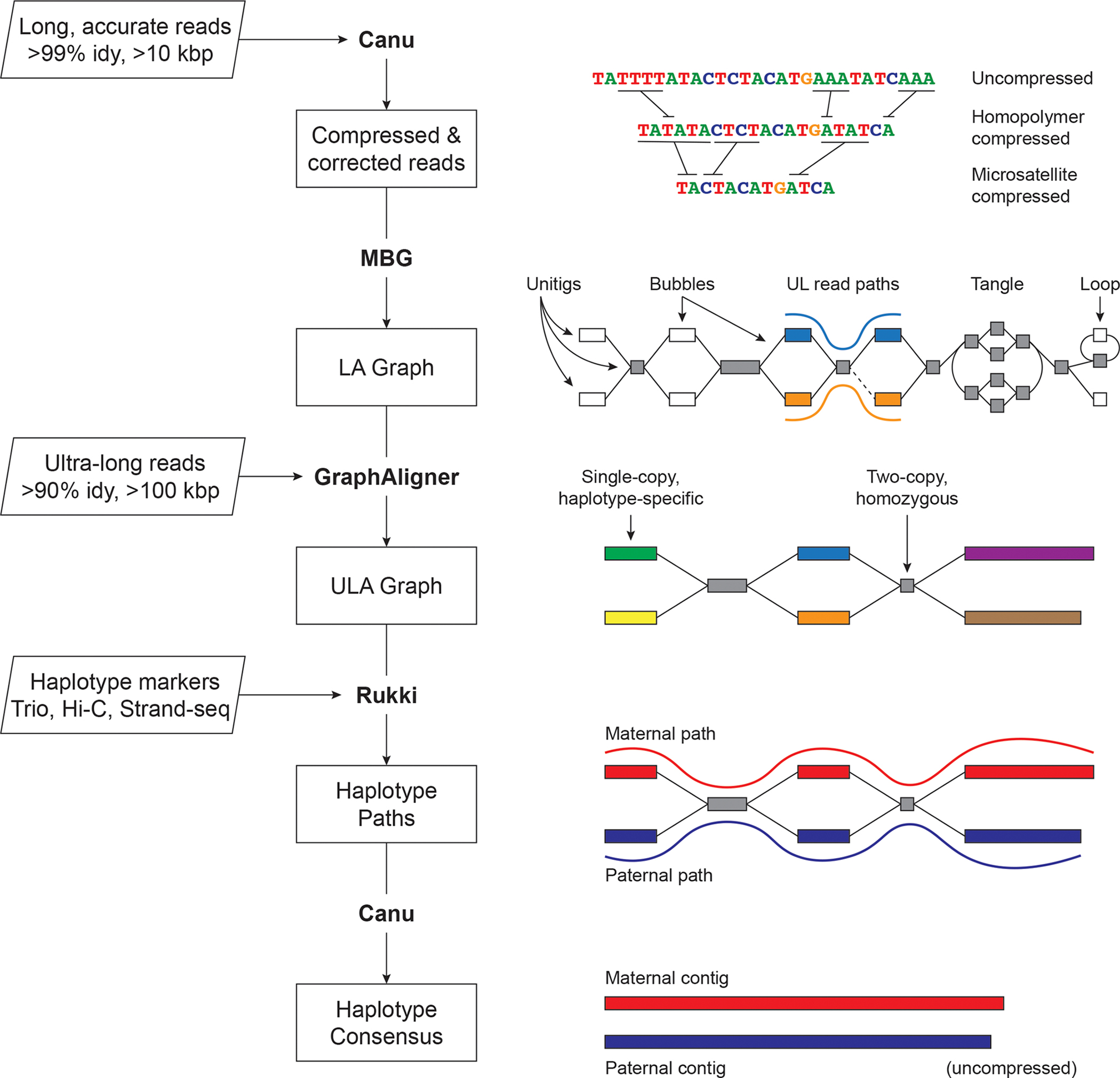
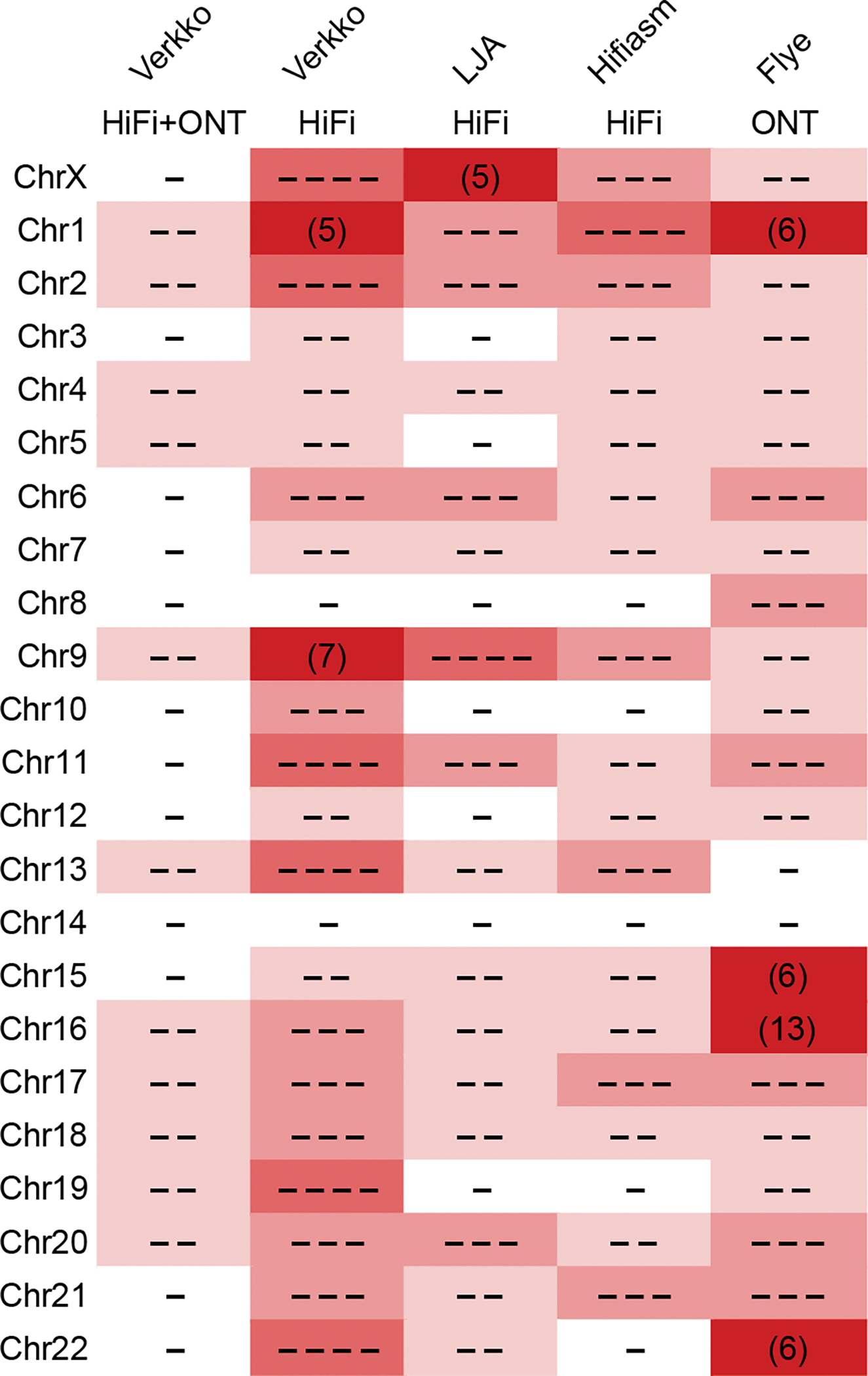
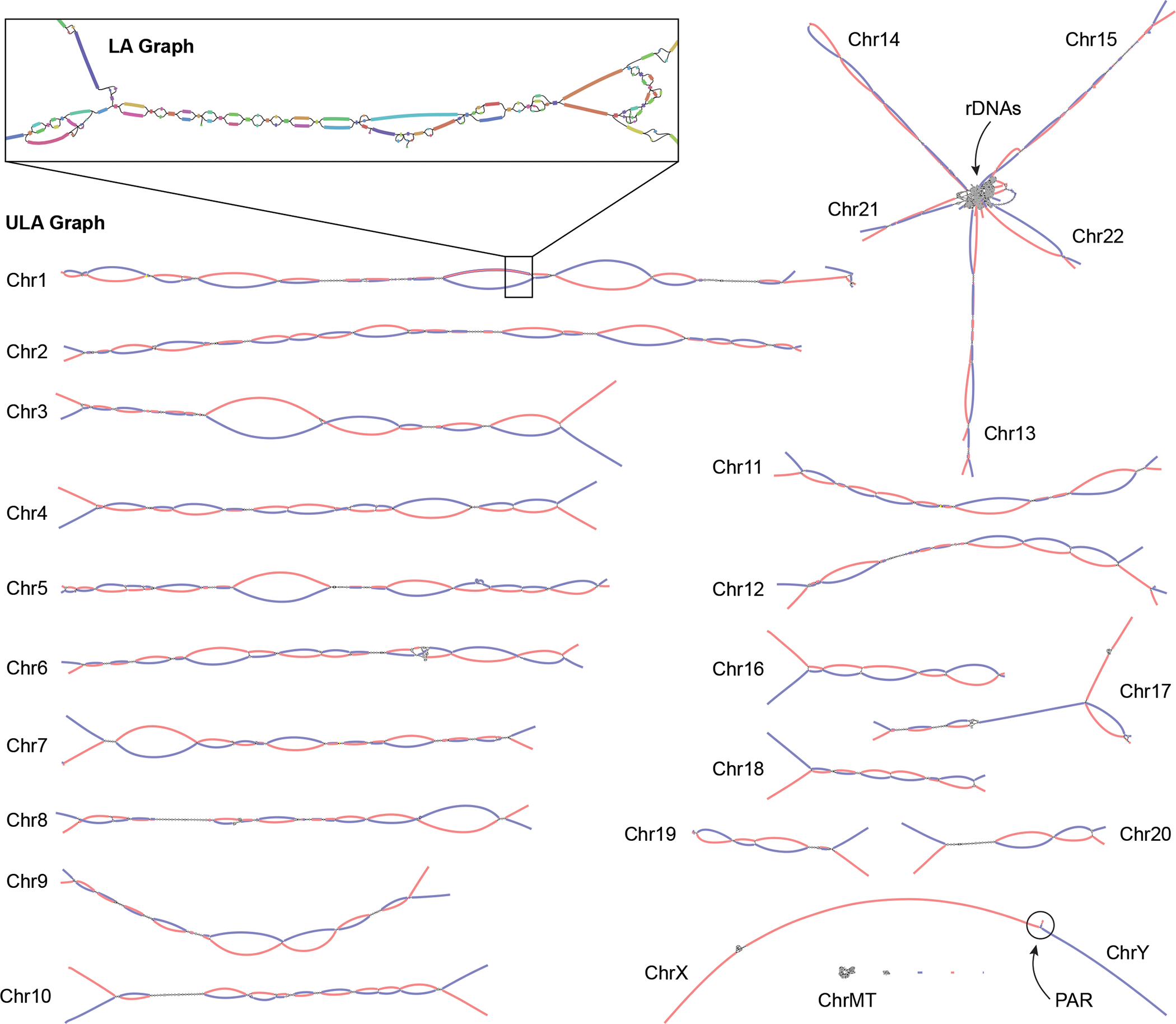
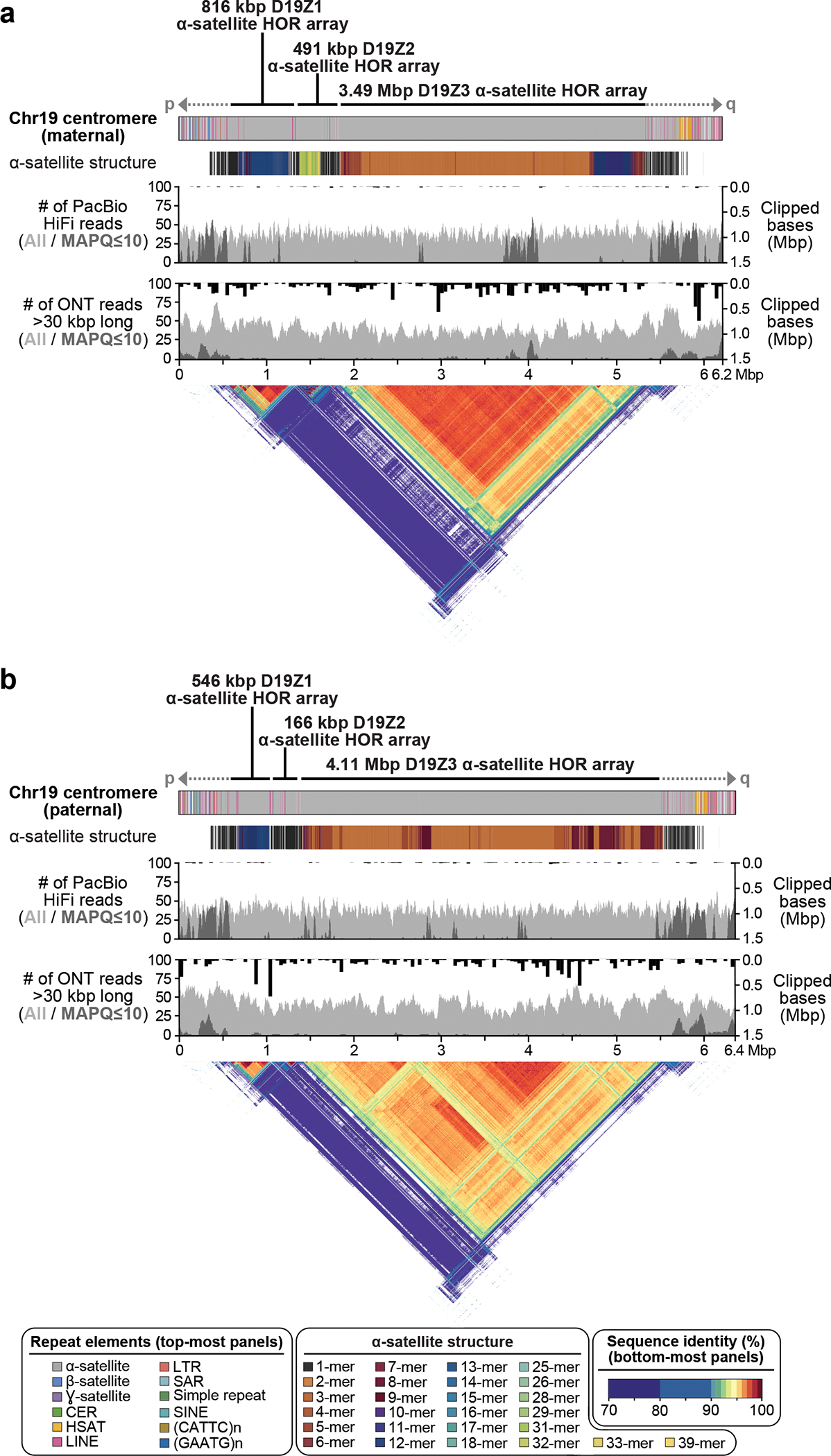
The Telomere-to-Telomere consortium recently assembled the first truly complete sequence of a human genome. To resolve the most complex repeats, this project relied on manual integration of ultra-long Oxford Nanopore sequencing reads with a high-resolution assembly graph built from long, accurate PacBio high-fidelity reads. We have improved and automated this strategy in Verkko, an iterative, graph-based pipeline for assembling complete, diploid genomes. Verkko begins with a multiplex de Bruijn graph built from long, accurate reads and progressively simplifies this graph by integrating ultra-long reads and haplotype-specific markers. The result is a phased, diploid assembly of both haplotypes, with many chromosomes automatically assembled from telomere to telomere. Running Verkko on the HG002 human genome resulted in 20 of 46 diploid chromosomes assembled without gaps at 99.9997% accuracy. The complete assembly of diploid genomes is a critical step towards the construction of comprehensive pangenome databases and chromosome-scale comparative genomics.
© 2023. This is a U.S. Government work and not under copyright protection in the US; foreign copyright protection may apply.
Conflict of interest statement
Competing interest declaration
EEE is on the scientific advisory board of DNAnexus, Inc. SK has received travel funds to speak at events hosted by Oxford Nanopore Technologies. SN is an employee of Oxford Nanopore Technologies. The remaining authors declare no competing interests.
Figures
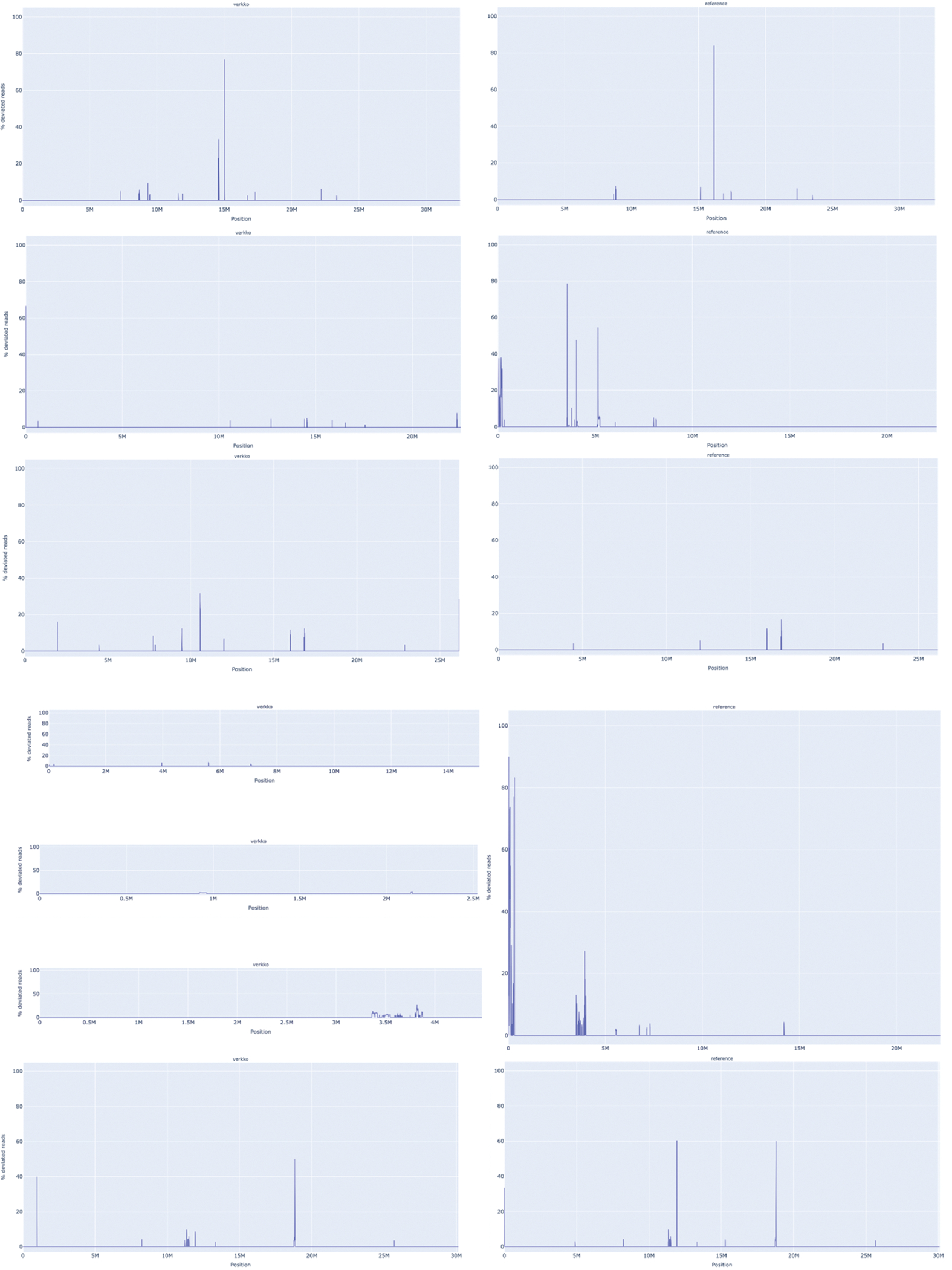
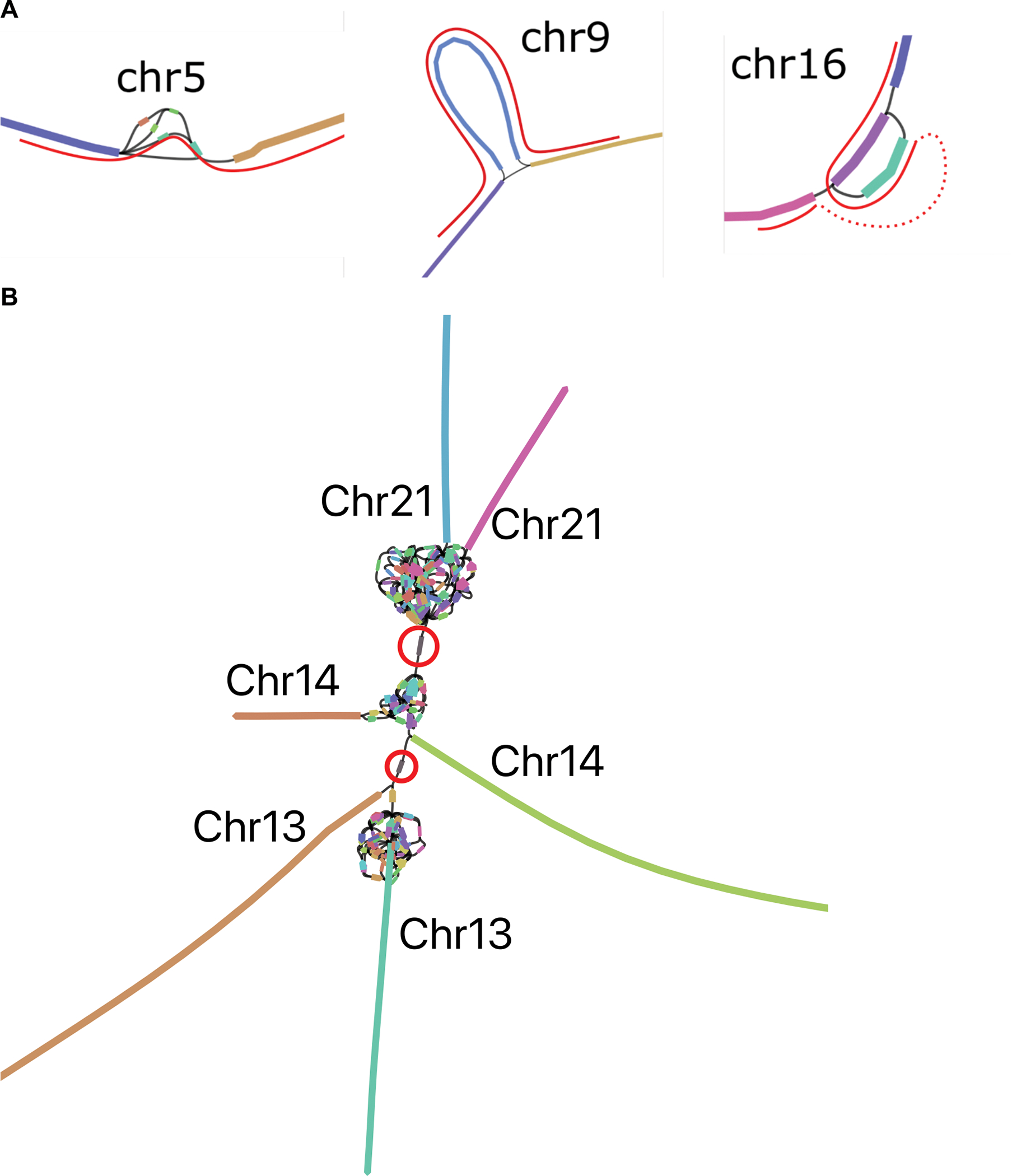
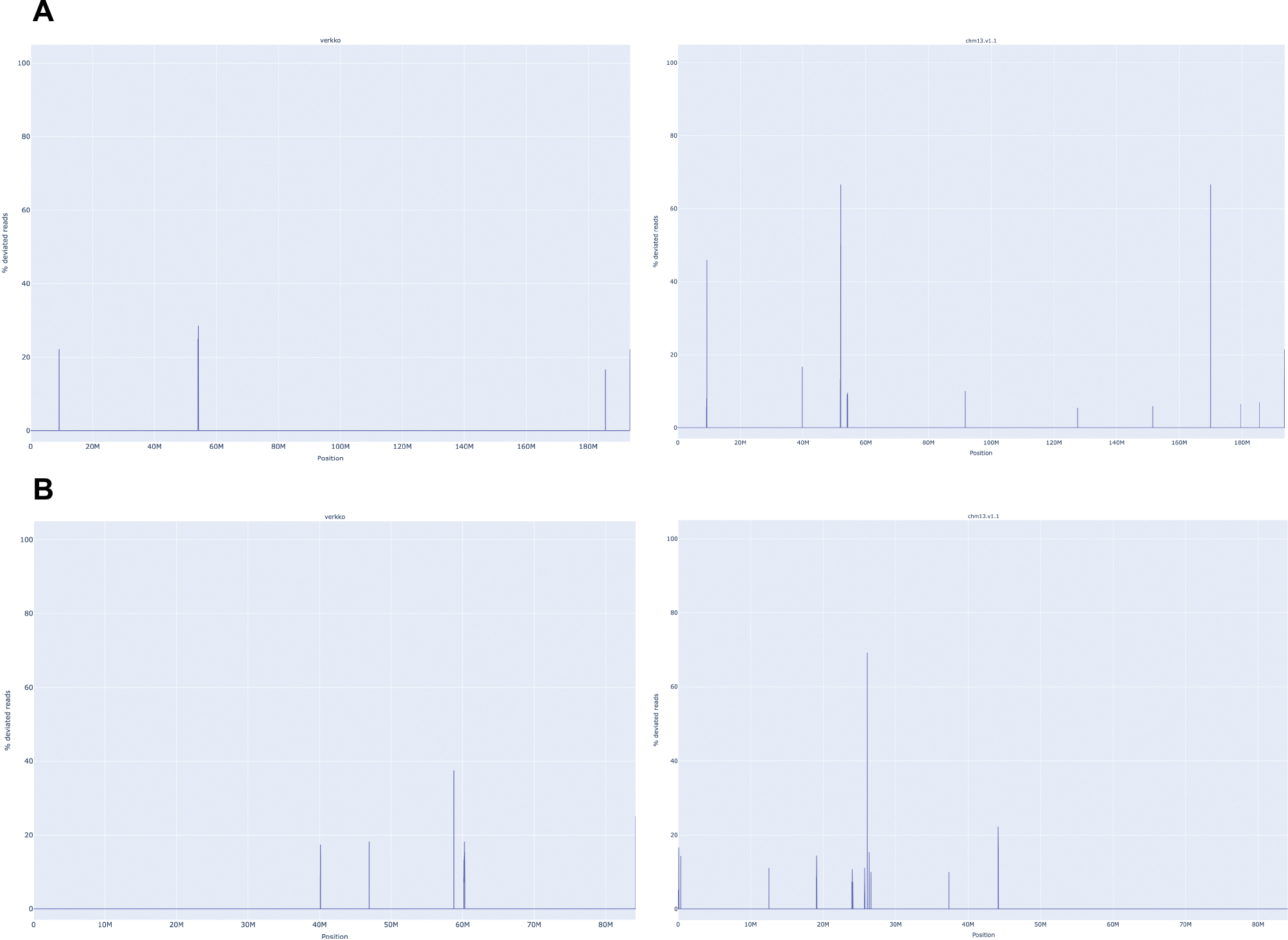
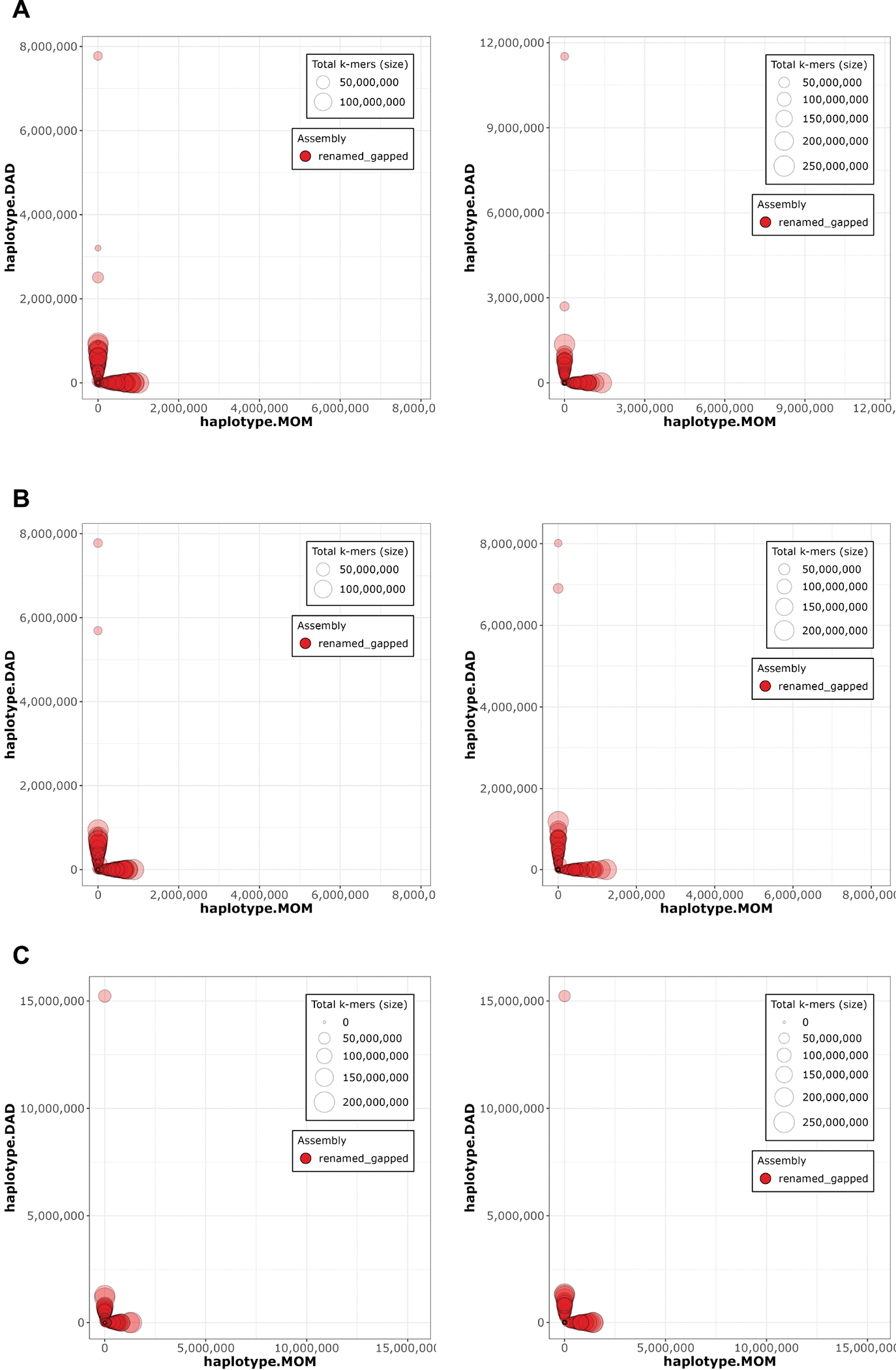
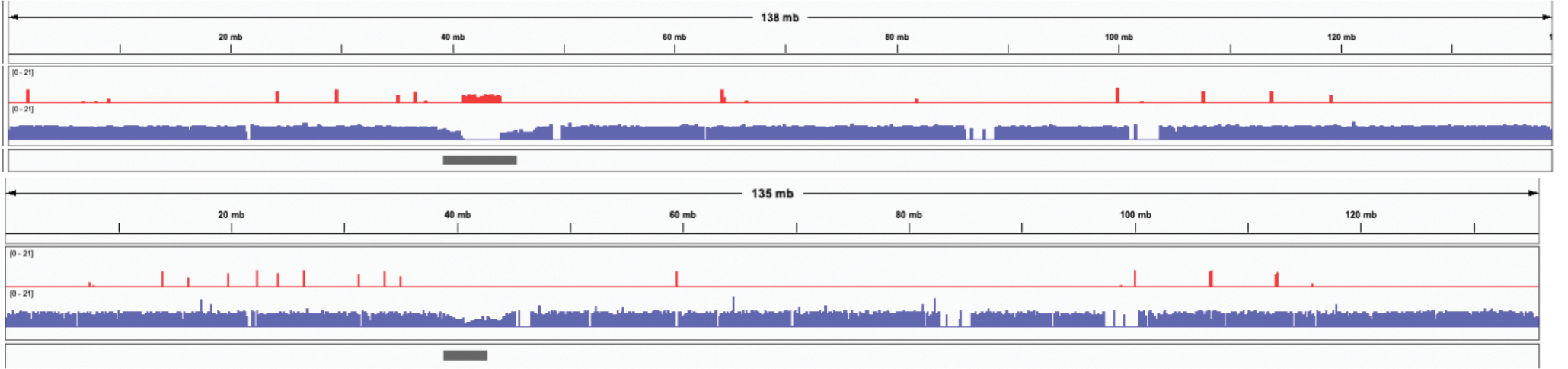
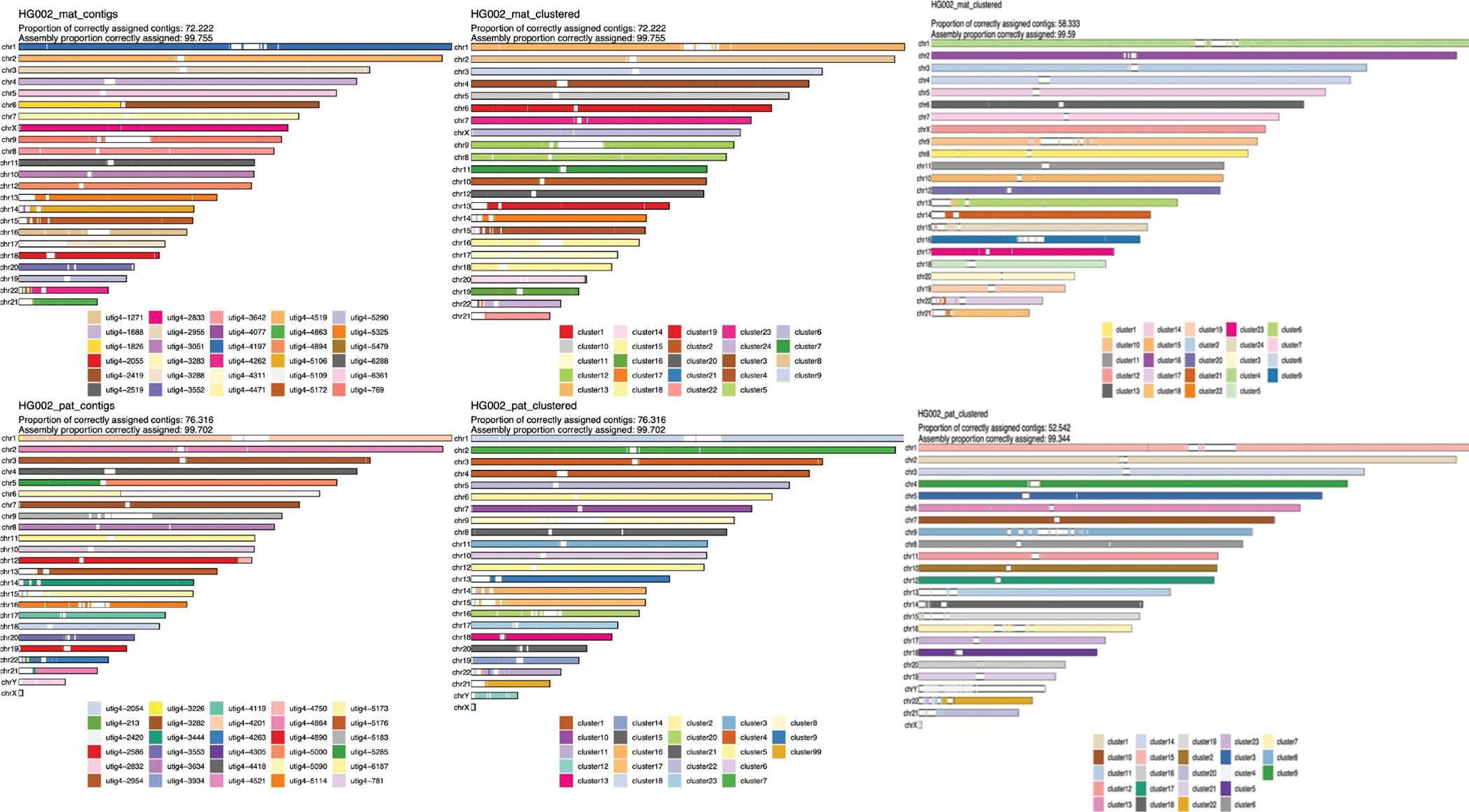
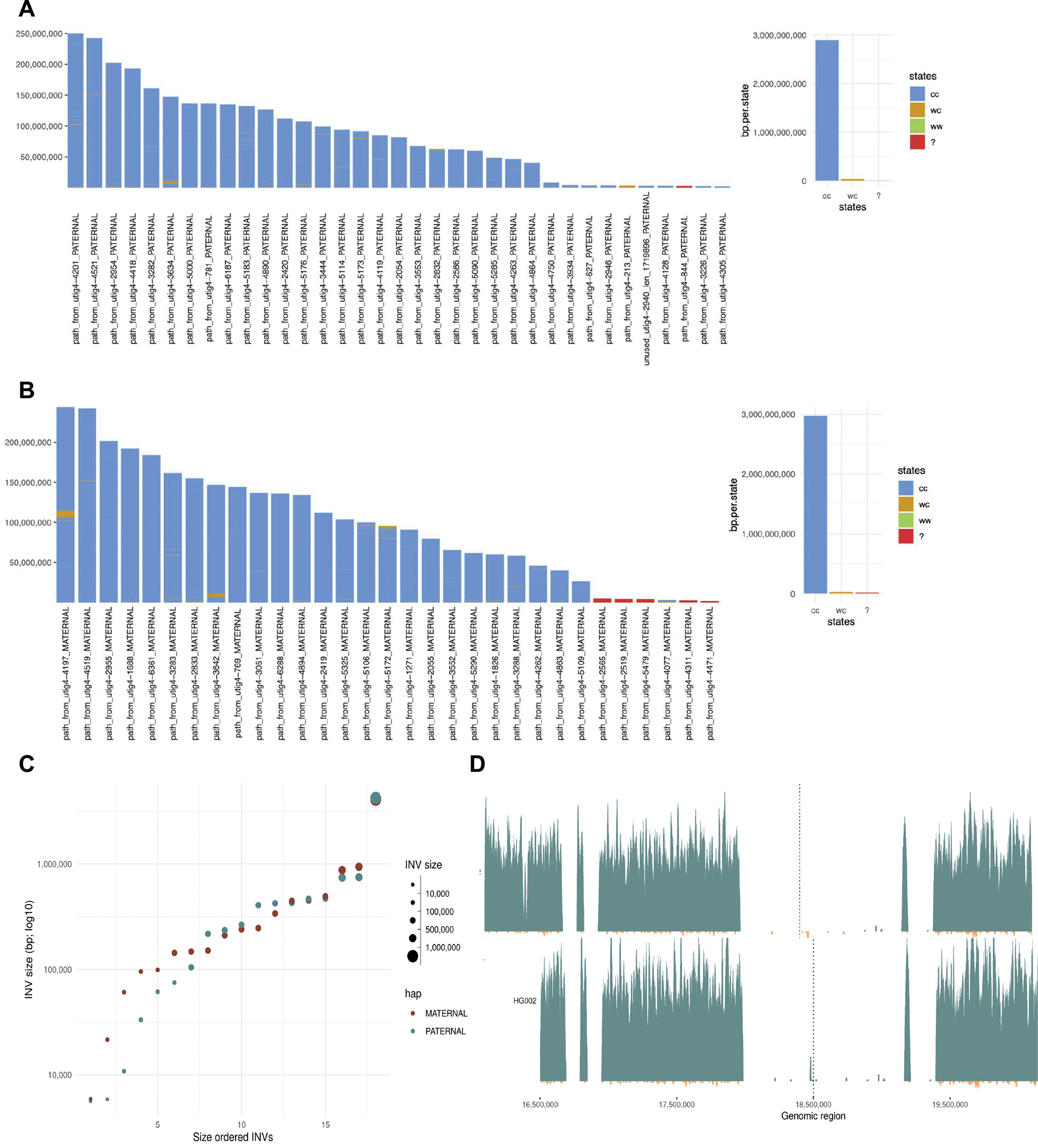
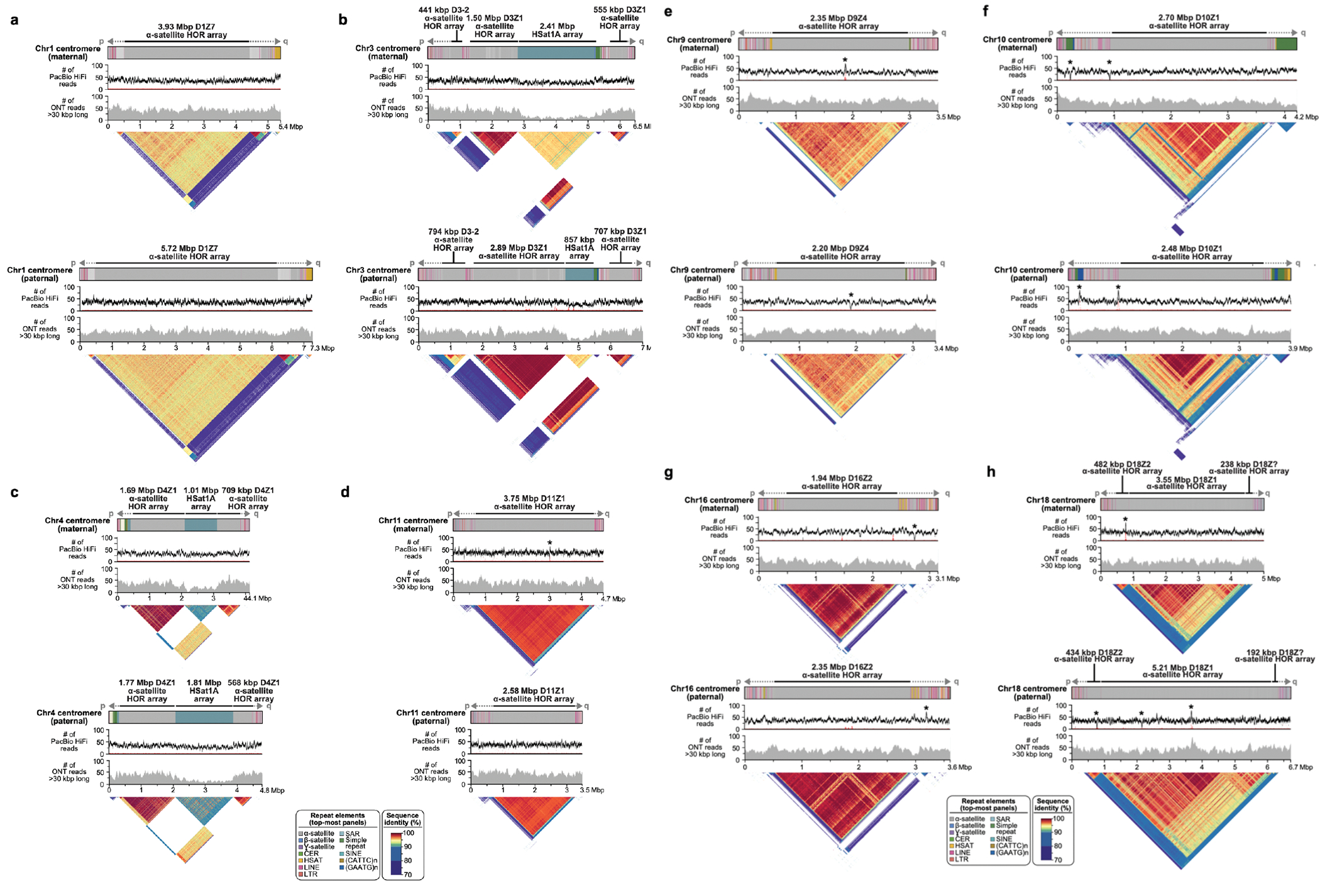
References
-
- Nagarajan N & Pop M Sequencing and genome assembly using next-generation technologies. Methods in molecular biology (Clifton, N.J.) vol. 673 1–17 (2010). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
