

This is a preprint.
CPA-Perturb-seq: Multiplexed single-cell characterization of alternative polyadenylation regulators
- PMID: 36798324
- PMCID: PMC9934614
- DOI: 10.1101/2023.02.09.527751
CPA-Perturb-seq: Multiplexed single-cell characterization of alternative polyadenylation regulators
Update in
-
Multiplexed single-cell characterization of alternative polyadenylation regulators.Cell. 2024 Aug 8;187(16):4408-4425.e23. doi: 10.1016/j.cell.2024.06.005. Epub 2024 Jun 25. Cell. 2024. PMID: 38925112 Free PMC article.
Abstract
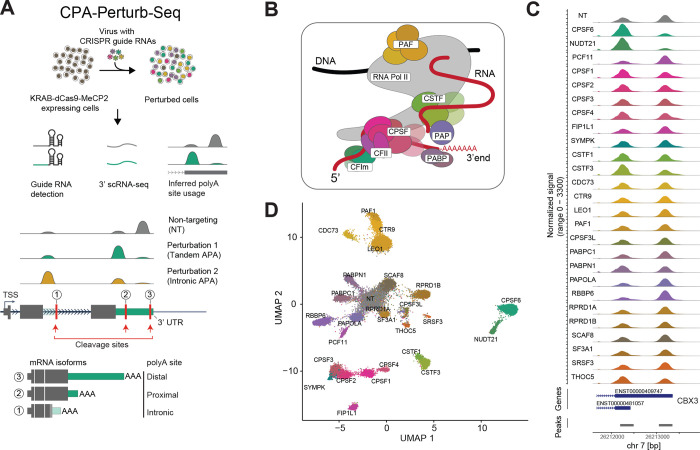
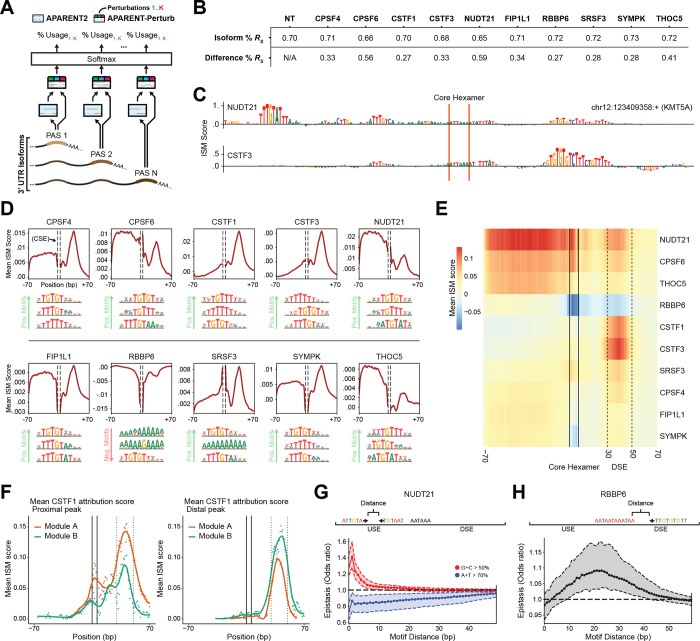
Most mammalian genes have multiple polyA sites, representing a substantial source of transcript diversity that is governed by the cleavage and polyadenylation (CPA) regulatory machinery. To better understand how these proteins govern polyA site choice we introduce CPA-Perturb-seq, a multiplexed perturbation screen dataset of 42 known CPA regulators with a 3' scRNA-seq readout that enables transcriptome-wide inference of polyA site usage. We develop a statistical framework to specifically identify perturbation-dependent changes in intronic and tandem polyadenylation, and discover modules of co-regulated polyA sites exhibiting distinct functional properties. By training a multi-task deep neural network (APARENT-Perturb) on our dataset, we delineate a cis-regulatory code that predicts responsiveness to perturbation and reveals interactions between distinct regulatory complexes. Finally, we leverage our framework to re-analyze published scRNA-seq datasets, identifying new regulators that affect the relative abundance of alternatively polyadenylated transcripts, and characterizing extensive cellular heterogeneity in 3' UTR length amongst antibody-producing cells. Our work highlights the potential for multiplexed single-cell perturbation screens to further our understanding of post-transcriptional regulation in vitro and in vivo.
Conflict of interest statement
COMPETING INTERESTS In the past three years, R.S. has worked as a consultant for Bristol-Myers Squibb, Regeneron, and Kallyope and served as an SAB member for ImmunAI, Resolve Biosciences, Nanostring, and the NYC Pandemic Response Lab. A.K. is on the SAB of PatchBio Inc., SerImmune Inc., AINovo Inc., TensorBio Inc., and OpenTargets; was a consultant with Illumina Inc. until Jan 2023; and owns shares in DeepGenomics Inc., Immunai Inc. and Freenome Inc. J.L. is an employee of Calico Life Sciences LLC as of 11/21/2022.
Figures




References
-
- Gruber A.J., and Zavolan M. (2019). Alternative cleavage and polyadenylation in health and disease. Nat. Rev. Genet. 20, 599–614. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources