

This is a preprint.
Scalable co-sequencing of RNA and DNA from individual nuclei
- PMID: 36798358
- PMCID: PMC9934633
- DOI: 10.1101/2023.02.09.527940
Scalable co-sequencing of RNA and DNA from individual nuclei
Update in
-
Scalable co-sequencing of RNA and DNA from individual nuclei.Nat Methods. 2025 Mar;22(3):477-487. doi: 10.1038/s41592-024-02579-x. Epub 2025 Feb 12. Nat Methods. 2025. PMID: 39939719 Free PMC article.
Abstract
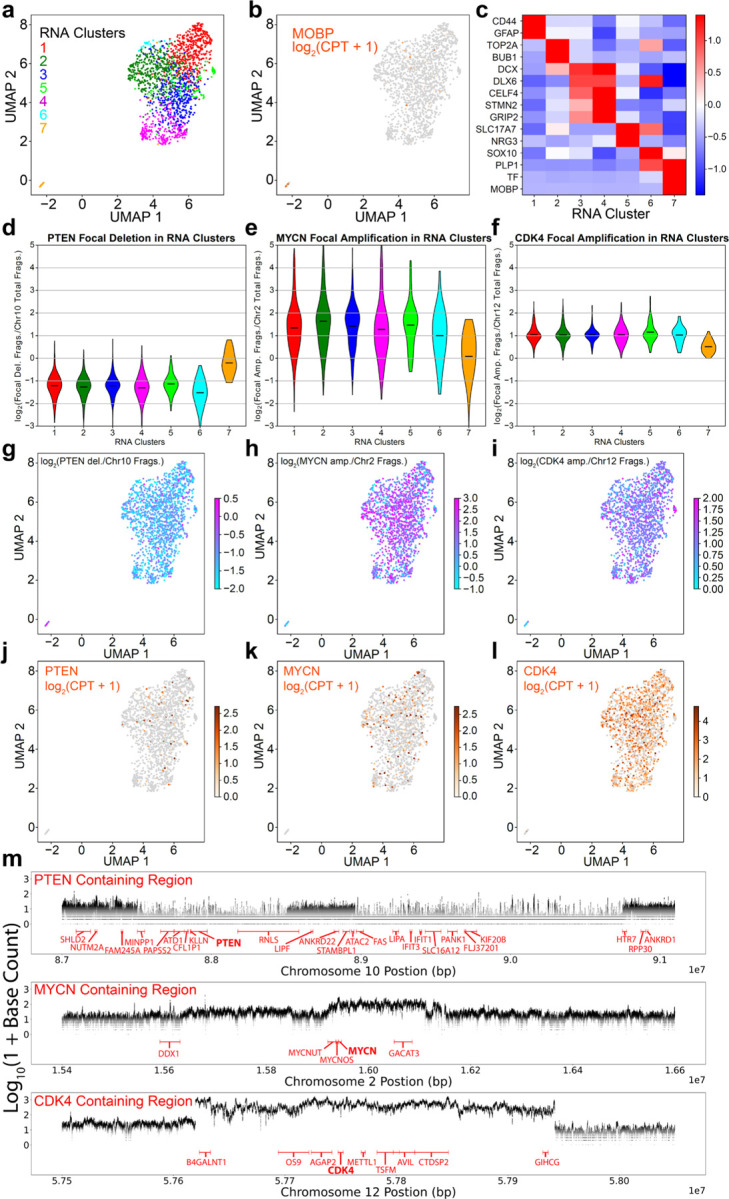
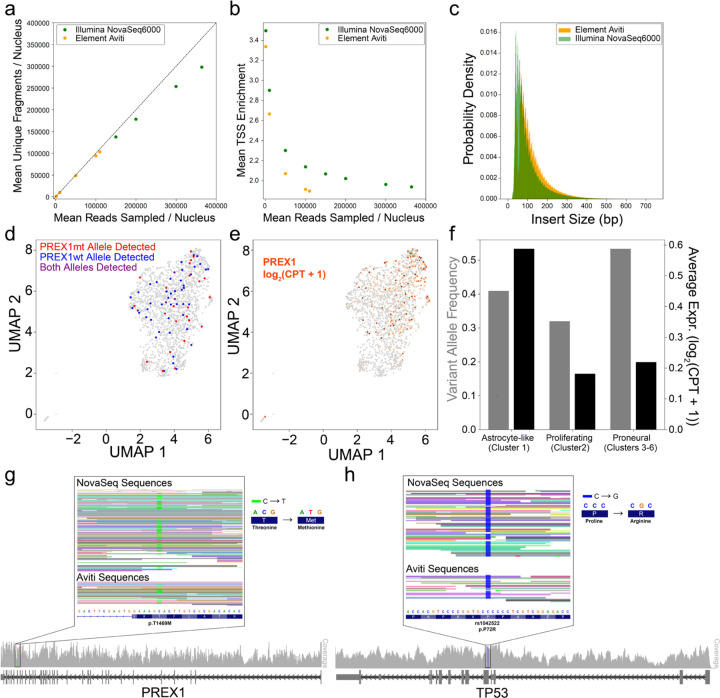
The ideal technology for directly investigating the relationship between genotype and phenotype would analyze both RNA and DNA genome-wide and with single-cell resolution. However, existing tools lack the throughput required for comprehensive analysis of complex tumors and tissues. We introduce a highly scalable method for jointly profiling DNA and expression following nucleosome depletion (DEFND-seq). In DEFND-seq, nuclei are nucleosome-depleted, tagmented, and separated into individual droplets for mRNA and genomic DNA barcoding. Once nuclei have been depleted of nucleosomes, subsequent steps can be performed using the widely available 10x Genomics droplet microfluidic technology and commercial kits without experimental modification. We demonstrate the production of high-complexity mRNA and gDNA sequencing libraries from thousands of individual nuclei from both cell lines and archived surgical specimens for associating gene expression phenotypes with both copy number and single nucleotide variants.
Conflict of interest statement
Competing Interests P.A.S. receives patent royalties from Guardant Health.
Figures
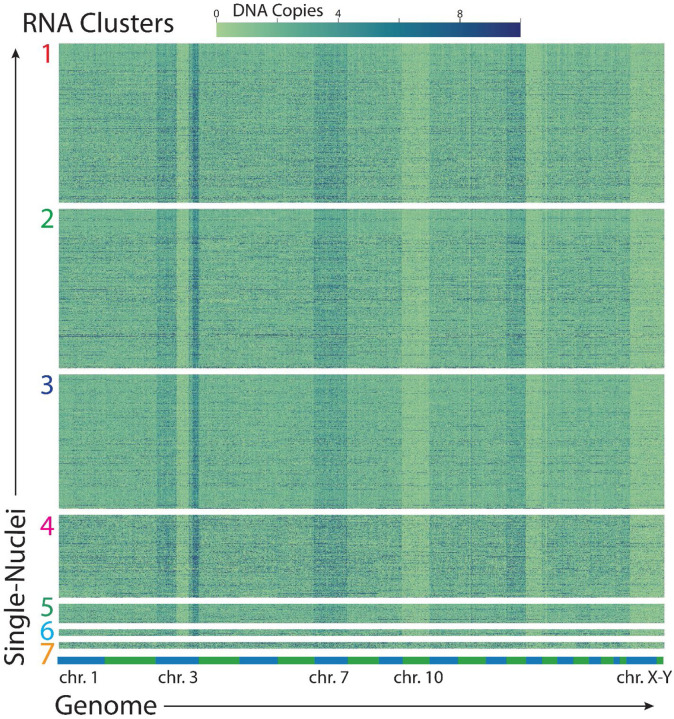
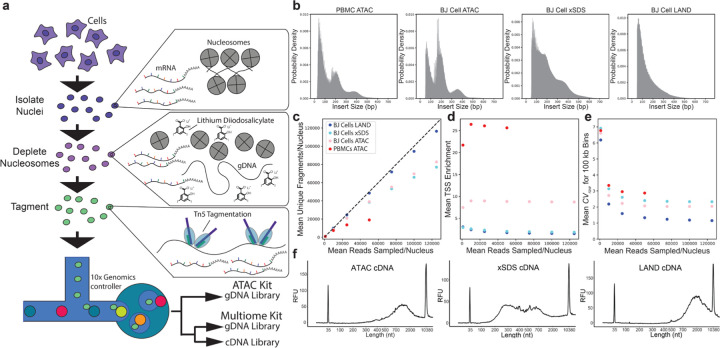
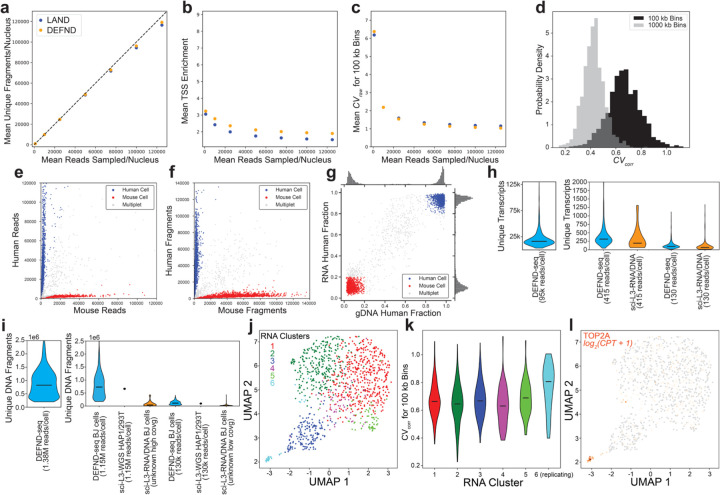
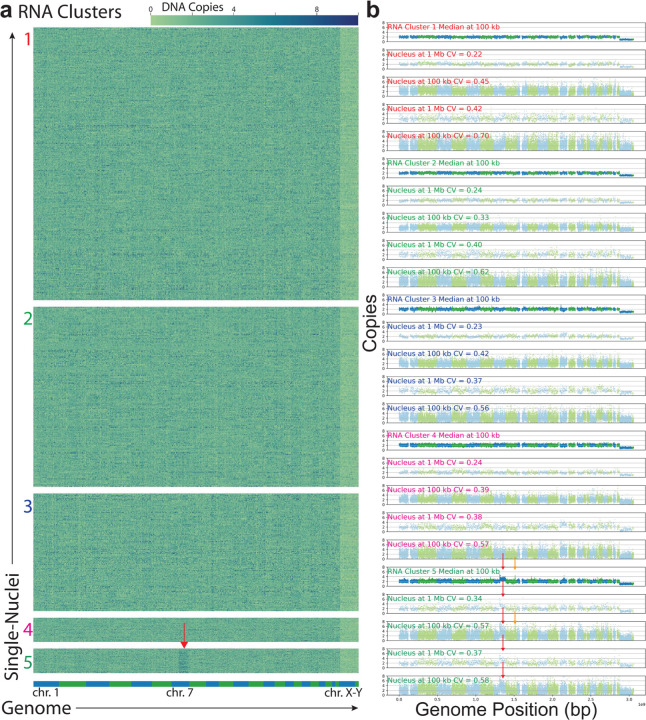
References
-
- Suvà M.L. & Tirosh I. Single-Cell RNA Sequencing in Cancer: Lessons Learned and Emerging Challenges. Mol. Cell 75, 7–12 (2019). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases