

A Kmer-based paired-end read de novo assembler and genotyper for canine MHC class I genotyping
- PMID: 36798440
- PMCID: PMC9926114
- DOI: 10.1016/j.isci.2023.105996
A Kmer-based paired-end read de novo assembler and genotyper for canine MHC class I genotyping
Abstract
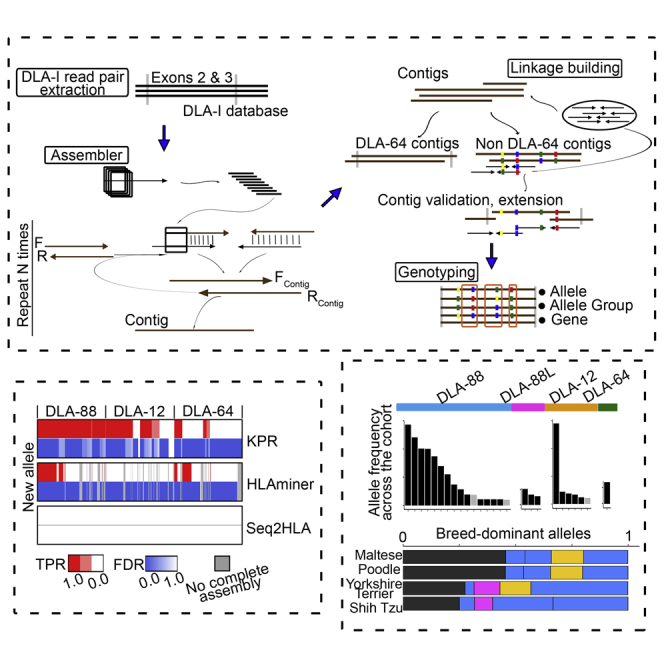
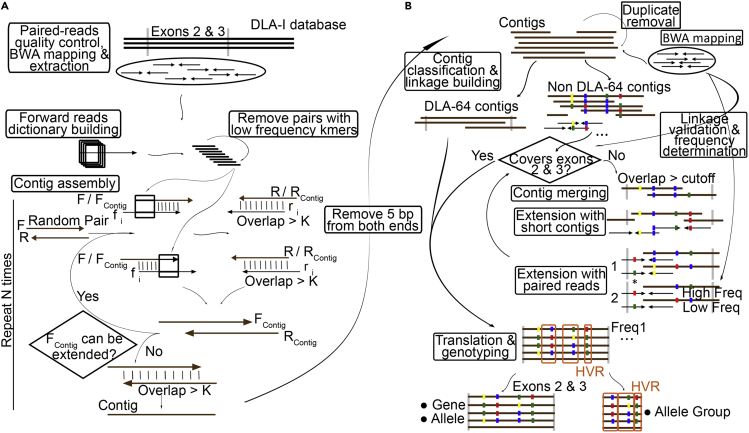
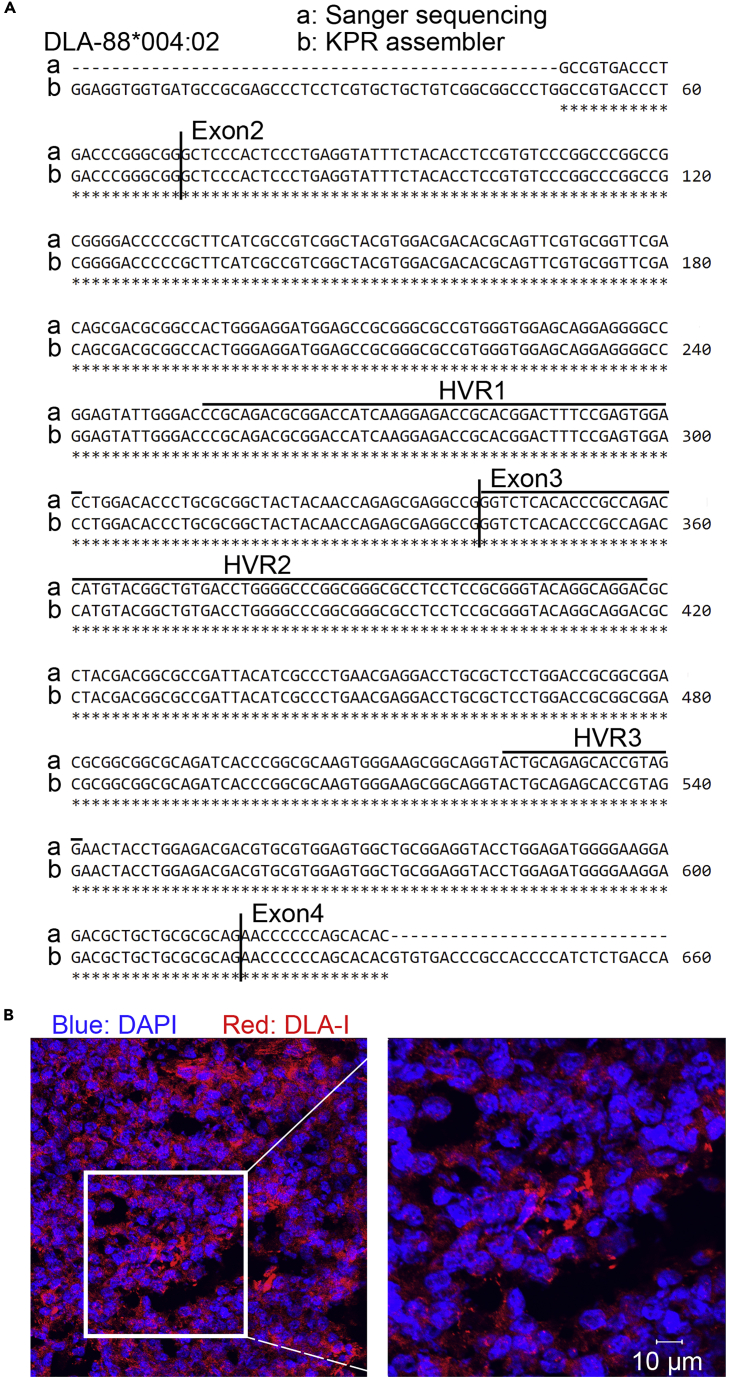
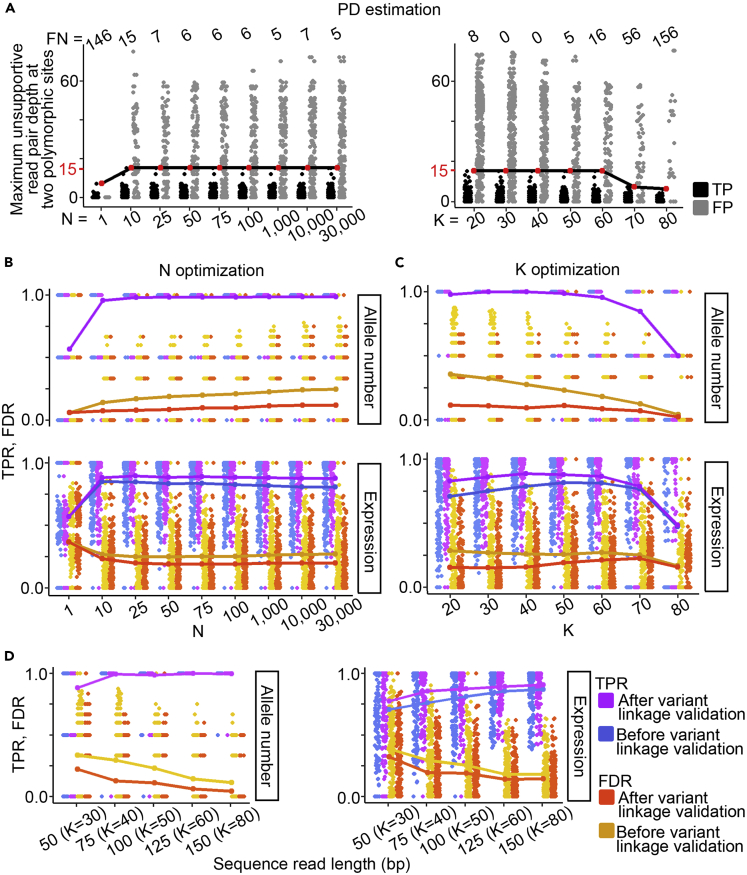
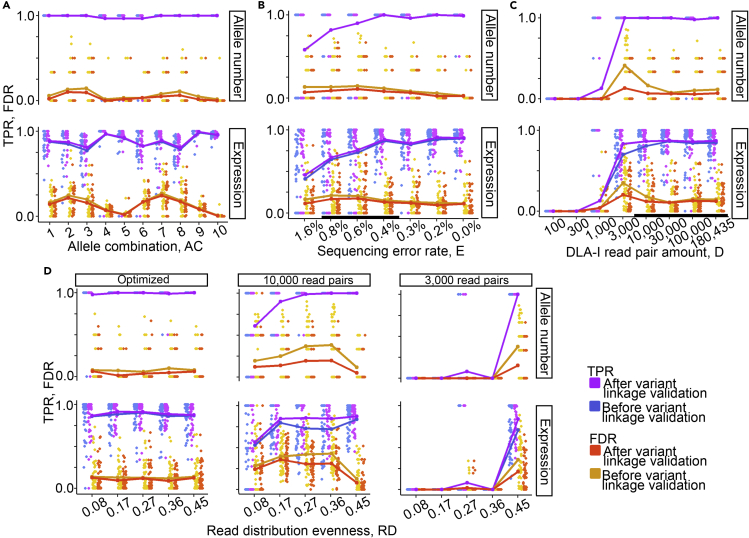
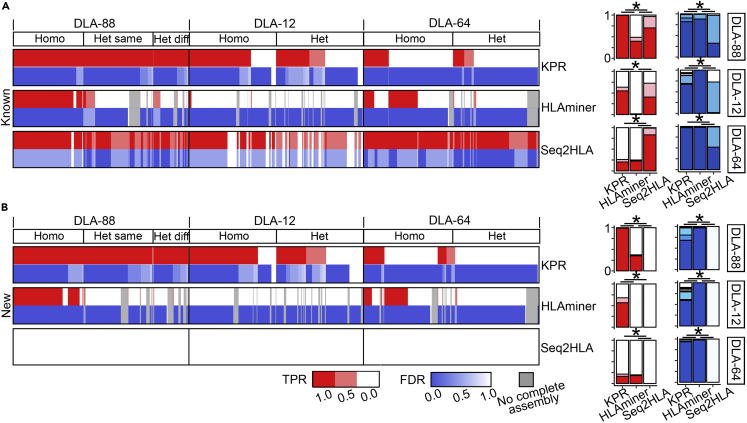
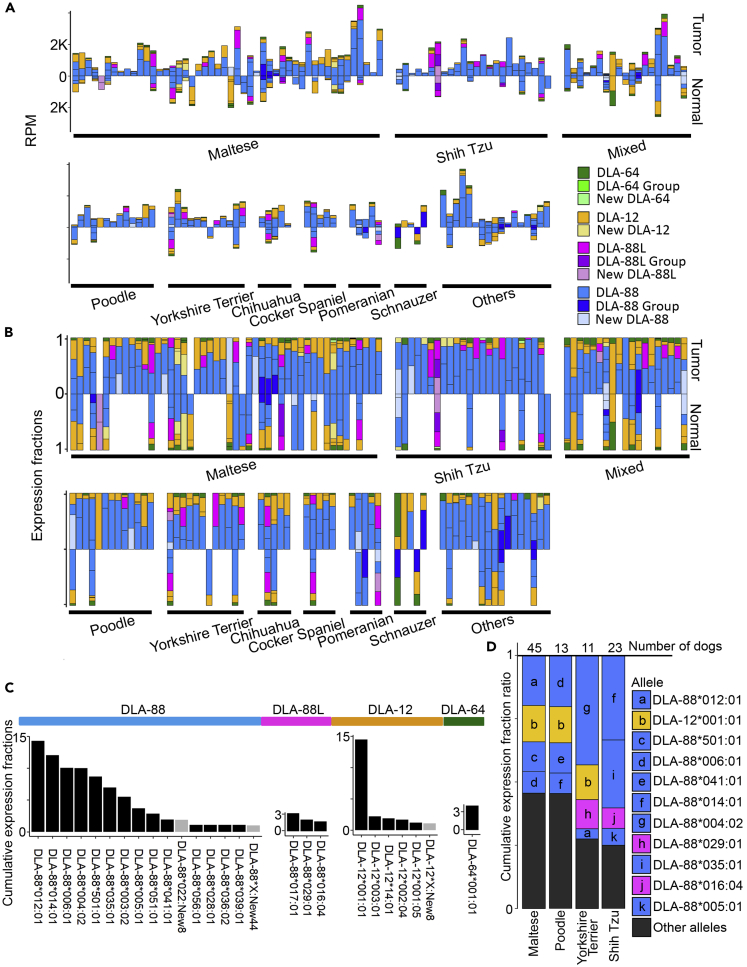
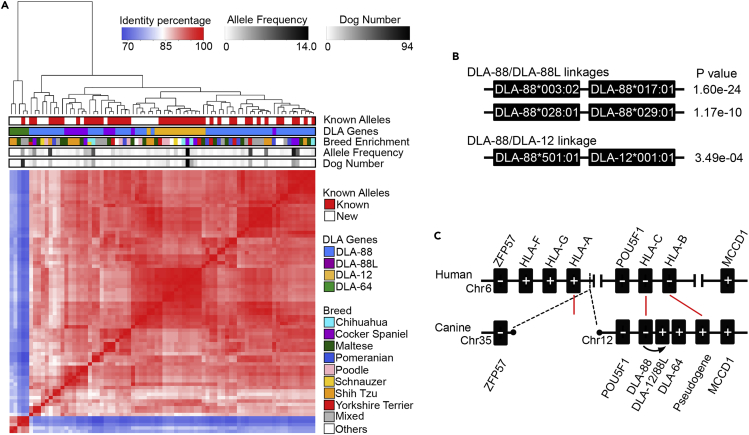
The major histocompatibility complex class I (MHC-I) genes are highly polymorphic. MHC-I genotyping is required for determining the peptide epitopes available to an individual's T-cell repertoire. Current genotyping software tools do not work for the dog, due to very limited known canine alleles. To address this, we developed a Kmer-based paired-end read (KPR) de novo assembler and genotyper, which assemble paired-end RNA-seq reads from MHC-I regions into contigs, and then genotype each contig and estimate its expression level. KPR tools outperform other popular software examined in typing new alleles. We used KPR tools to successfully genotype152 dogs from a published dataset. The study discovers 33 putative new alleles, finds dominant alleles in 4 dog breeds, and builds allele diversity and expression landscapes among the 152 dogs. Our software meets a significant need in biomedical research.
Keywords: Biocomputational method; Computational bioinformatics; Genomic analysis.
© 2023 The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- London C.A., Acquaviva J., Smith D.L., Sequeira M., Ogawa L.S., Gardner H.L., Bernabe L.F., Bear M.D., Bechtel S.A., Proia D.A. Consecutive day hsp90 inhibitor administration improves efficacy in murine models of kit-driven malignancies and canine mast cell tumors. Clin. Cancer Res. 2018;24:6396–6407. doi: 10.1158/1078-0432.CCR-18-0703. - DOI - PMC - PubMed
-
- Regan D.P., Chow L., Das S., Haines L., Palmer E., Kurihara J.N., Coy J.W., Mathias A., Thamm D.H., Gustafson D.L., Dow S.W. Losartan blocks osteosarcoma-elicited monocyte recruitment, and combined with the kinase inhibitor toceranib, exerts significant clinical benefit in canine metastatic osteosarcoma. Clin. Cancer Res. 2022;28:662–676. doi: 10.1158/1078-0432.CCR-21-2105. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
