

PlantTribes2: Tools for comparative gene family analysis in plant genomics
- PMID: 36798801
- PMCID: PMC9928214
- DOI: 10.3389/fpls.2022.1011199
PlantTribes2: Tools for comparative gene family analysis in plant genomics
Abstract
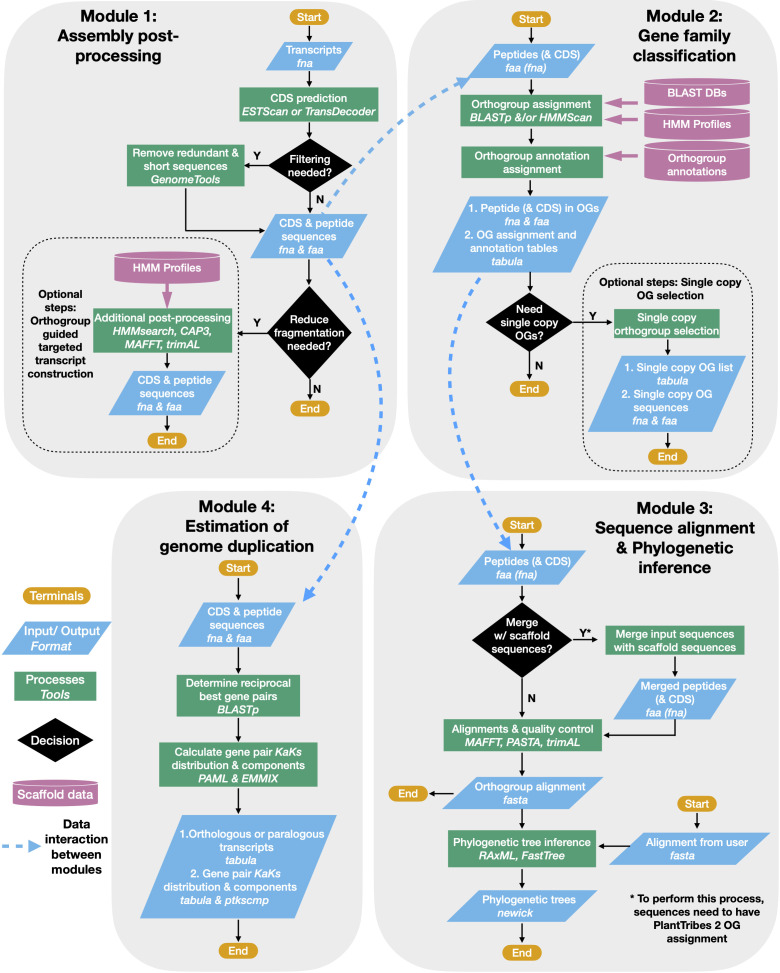
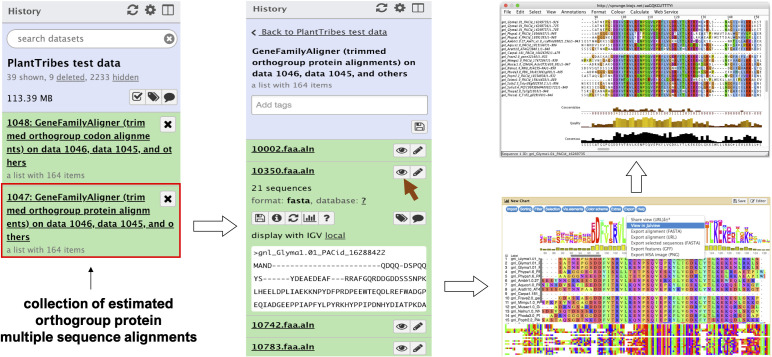
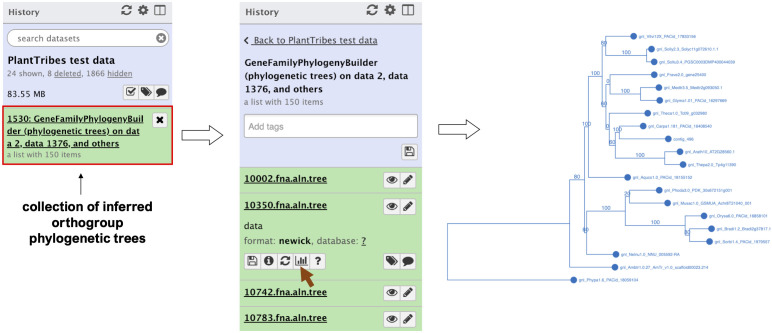
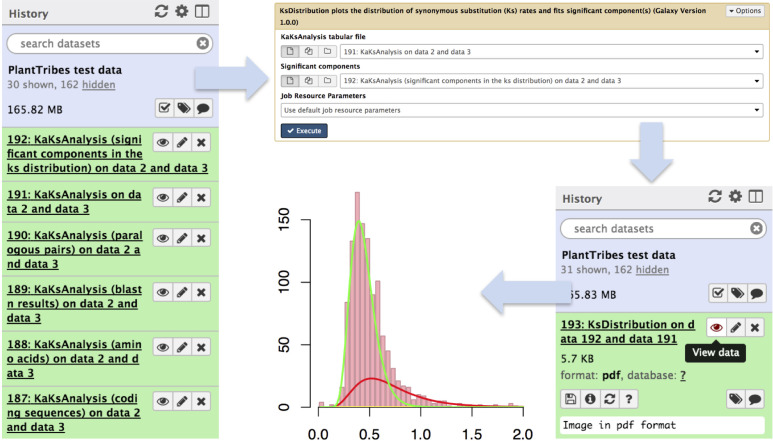
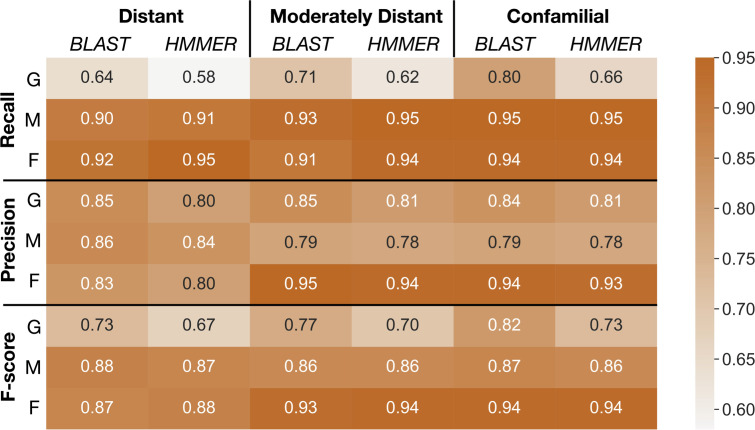
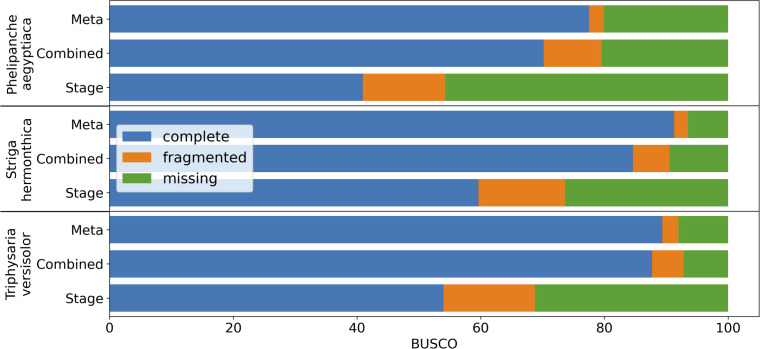
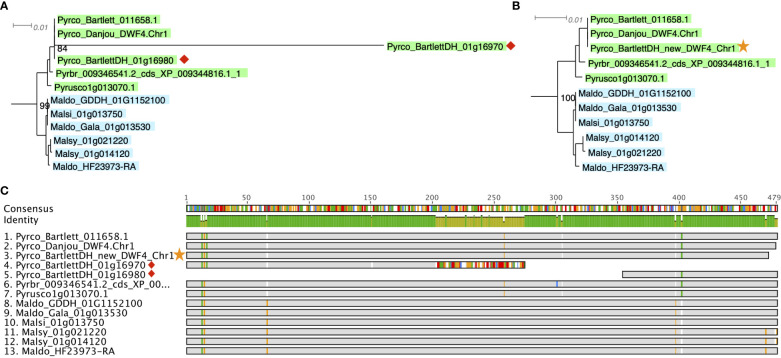
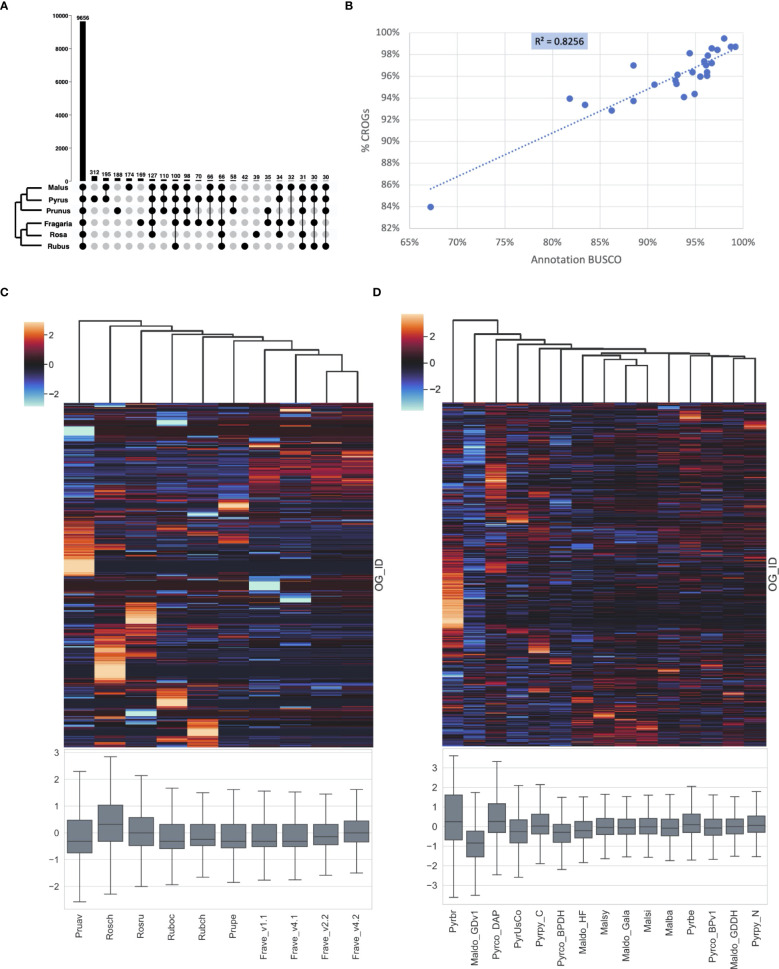
Plant genome-scale resources are being generated at an increasing rate as sequencing technologies continue to improve and raw data costs continue to fall; however, the cost of downstream analyses remains large. This has resulted in a considerable range of genome assembly and annotation qualities across plant genomes due to their varying sizes, complexity, and the technology used for the assembly and annotation. To effectively work across genomes, researchers increasingly rely on comparative genomic approaches that integrate across plant community resources and data types. Such efforts have aided the genome annotation process and yielded novel insights into the evolutionary history of genomes and gene families, including complex non-model organisms. The essential tools to achieve these insights rely on gene family analysis at a genome-scale, but they are not well integrated for rapid analysis of new data, and the learning curve can be steep. Here we present PlantTribes2, a scalable, easily accessible, highly customizable, and broadly applicable gene family analysis framework with multiple entry points including user provided data. It uses objective classifications of annotated protein sequences from existing, high-quality plant genomes for comparative and evolutionary studies. PlantTribes2 can improve transcript models and then sort them, either genome-scale annotations or individual gene coding sequences, into pre-computed orthologous gene family clusters with rich functional annotation information. Then, for gene families of interest, PlantTribes2 performs downstream analyses and customizable visualizations including, (1) multiple sequence alignment, (2) gene family phylogeny, (3) estimation of synonymous and non-synonymous substitution rates among homologous sequences, and (4) inference of large-scale duplication events. We give examples of PlantTribes2 applications in functional genomic studies of economically important plant families, namely transcriptomics in the weedy Orobanchaceae and a core orthogroup analysis (CROG) in Rosaceae. PlantTribes2 is freely available for use within the main public Galaxy instance and can be downloaded from GitHub or Bioconda. Importantly, PlantTribes2 can be readily adapted for use with genomic and transcriptomic data from any kind of organism.
Keywords: CROG analysis; applied agriculture; comparative genomics; galaxy; gene family phylogenetics; genome duplication; modular tools; multiple sequence alignment.
Copyright © 2023 Wafula, Zhang, Von Kuster, Leebens-Mack, Honaas and dePamphilis.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures

References
LinkOut - more resources
Full Text Sources
Other Literature Sources