

MET exon 14 skipping mutation is a hepatocyte growth factor (HGF)-dependent oncogenic driver in vitro and in humanised HGF knock-in mice
- PMID: 36799689
- PMCID: PMC10620121
- DOI: 10.1002/1878-0261.13397
MET exon 14 skipping mutation is a hepatocyte growth factor (HGF)-dependent oncogenic driver in vitro and in humanised HGF knock-in mice
Abstract
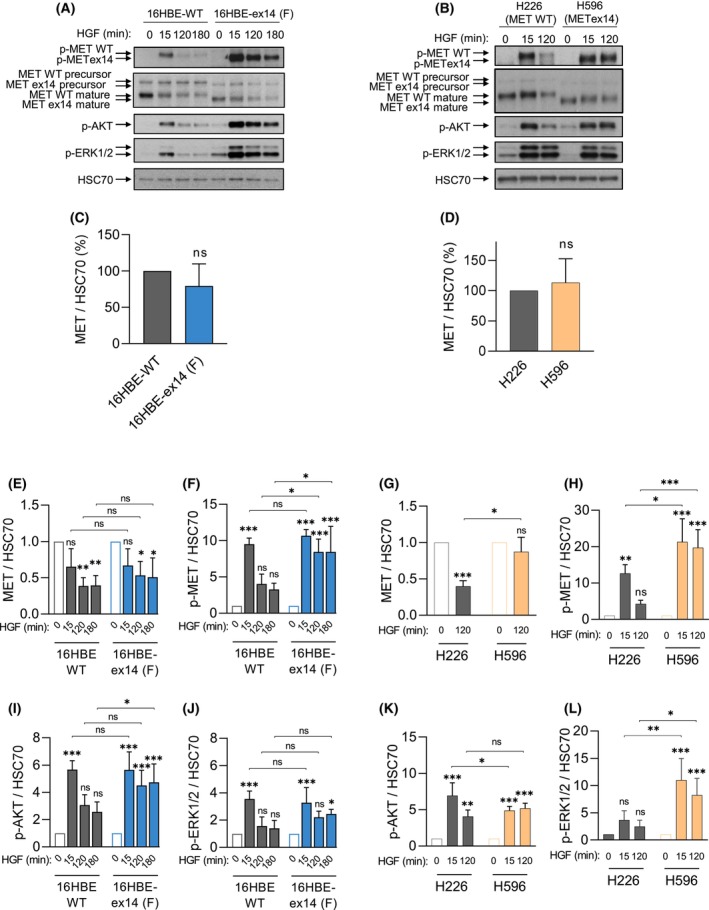
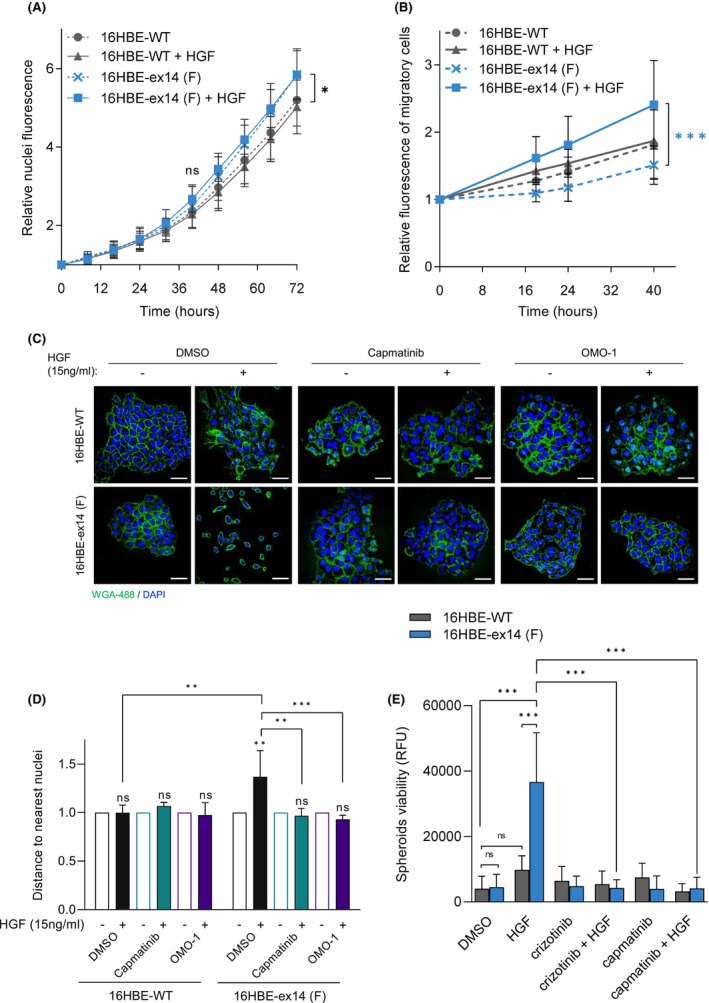
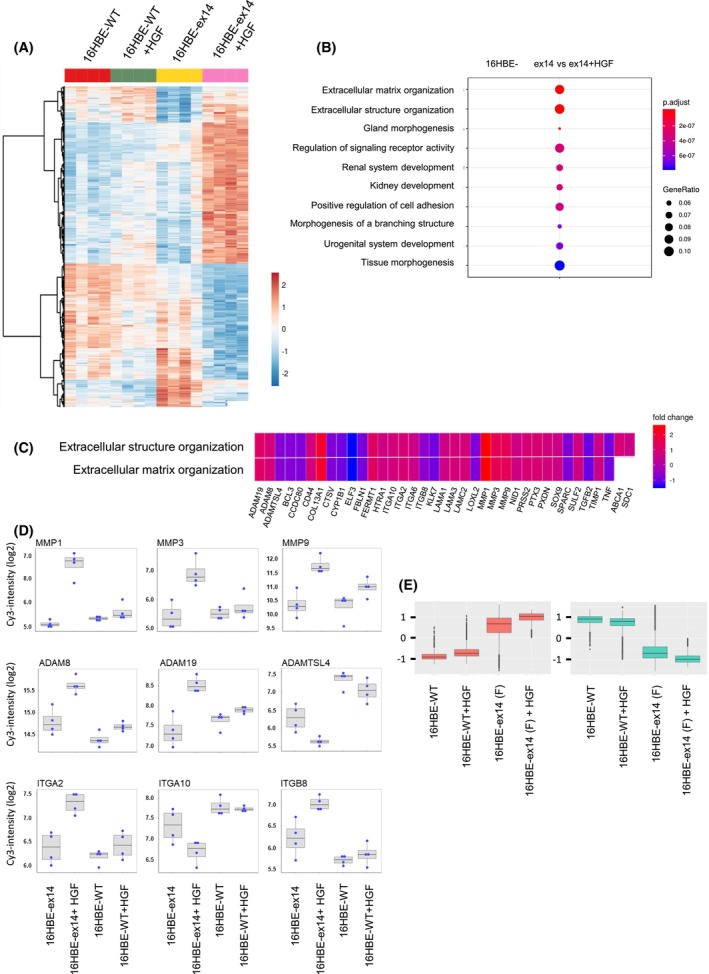
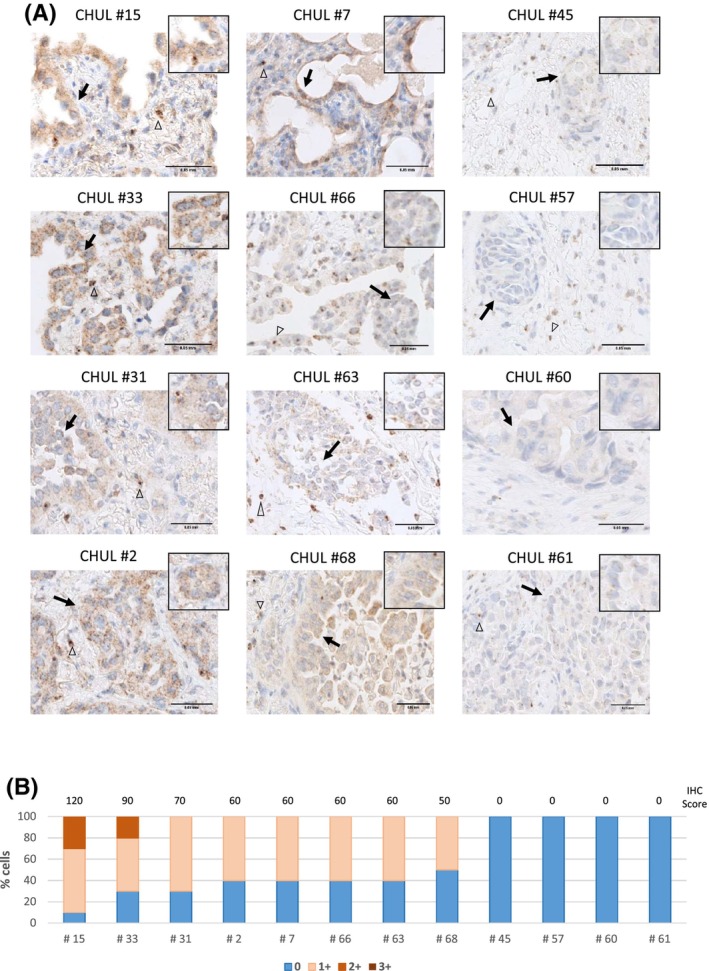
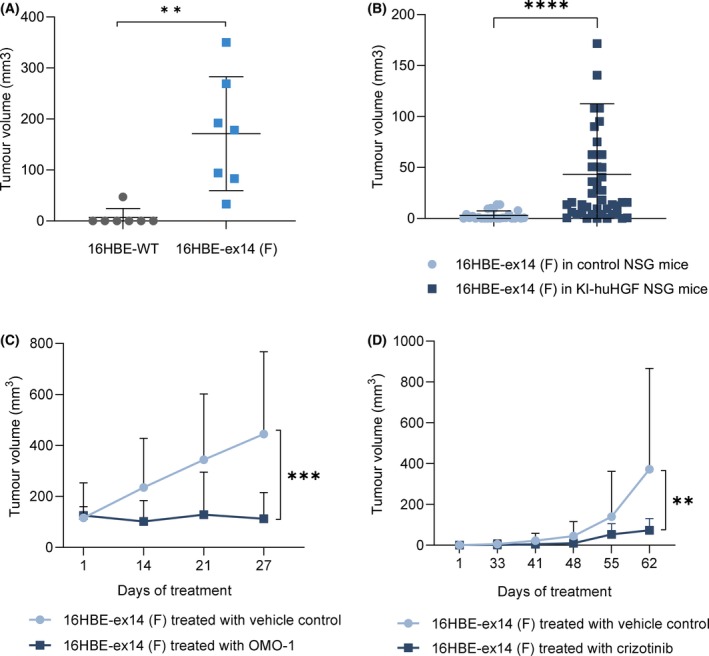
Exon skipping mutations of the MET receptor tyrosine kinase (METex14), increasingly reported in cancers, occur in 3-4% of non-small-cell lung cancer (NSCLC). Only 50% of patients have a beneficial response to treatment with MET-tyrosine kinase inhibitors (TKIs), underlying the need to understand the mechanism of METex14 oncogenicity and sensitivity to TKIs. Whether METex14 is a driver mutation and whether it requires hepatocyte growth factor (HGF) for its oncogenicity in a range of in vitro functions and in vivo has not been fully elucidated from previous preclinical models. Using CRISPR/Cas9, we developed a METex14/WT isogenic model in nontransformed human lung cells and report that the METex14 single alteration was sufficient to drive MET-dependent in vitro anchorage-independent survival and motility and in vivo tumorigenesis, sensitising tumours to MET-TKIs. However, we also show that human HGF (hHGF) is required, as demonstrated in vivo using a humanised HGF knock-in strain of mice and further detected in tumour cells of METex14 NSCLC patient samples. Our results also suggest that METex14 oncogenicity is not a consequence of an escape from degradation in our cell model. Thus, we developed a valuable model for preclinical studies and present results that have potential clinical implication.
Keywords: hepatocyte growth factor; lung cancer; preclinical models; targeted therapies; transcriptomic; tyrosine kinase receptor.
© 2023 The Authors. Molecular Oncology published by John Wiley & Sons Ltd on behalf of Federation of European Biochemical Societies.
Conflict of interest statement
PJ reports personal fees and non‐financial support from Novartis and Boehringer Ingelheim FRANCE, non‐financial support from Pierre Fabre, Chugai Pharma and MYLAN MEDICAL SAS, outside the submitted work. SH participates in advisory board for Brystol‐Myers Squibb and Boehringer Ingelheim. CD reports personal fees and non‐financial support from AstraZeneca, Novartis pharma SAS, Roche SAS, Boehringer Ingelheim France, Pfizer, outside the submitted work. MCC participates in advisory boards for Pfizer and Roche. ABC participated in advisory boards or received honoraria from Abbvie, Amgen, Astra‐Zeneca, Bristol‐Myers Squibb, Merck & Co, Pfizer, Roche, Novartis, Takeda, Janssen, Sanofi and received grants payed to ABC's institution from Novartis, Merck, Roche. TP headed OCTIMET Oncology NV and DeuterOncology NV. MFe, BH, SP, VG, AV, BDL, AM, GW, PG, JPM, SS, EC, JS, TS, PF, LG, MFi, DT, SK and ZK declare no competing financial interests.
Figures
References
-
- Bedard PL, Hyman DM, Davids MS, Siu LL. Small molecules, big impact: 20 years of targeted therapy in oncology. Lancet. 2020;395:1078–88. - PubMed
-
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non‐small‐cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39. - PubMed
-
- Liu X, Jia Y, Stoopler MB, Shen Y, Cheng H, Chen J, et al. Next‐generation sequencing of pulmonary Sarcomatoid carcinoma reveals high frequency of actionable MET gene mutations. J Clin Oncol. 2016;34:794–802. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
