

Variants in DTNA cause a mild, dominantly inherited muscular dystrophy
- PMID: 36799992
- PMCID: PMC10923638
- DOI: 10.1007/s00401-023-02551-7
Variants in DTNA cause a mild, dominantly inherited muscular dystrophy
Abstract
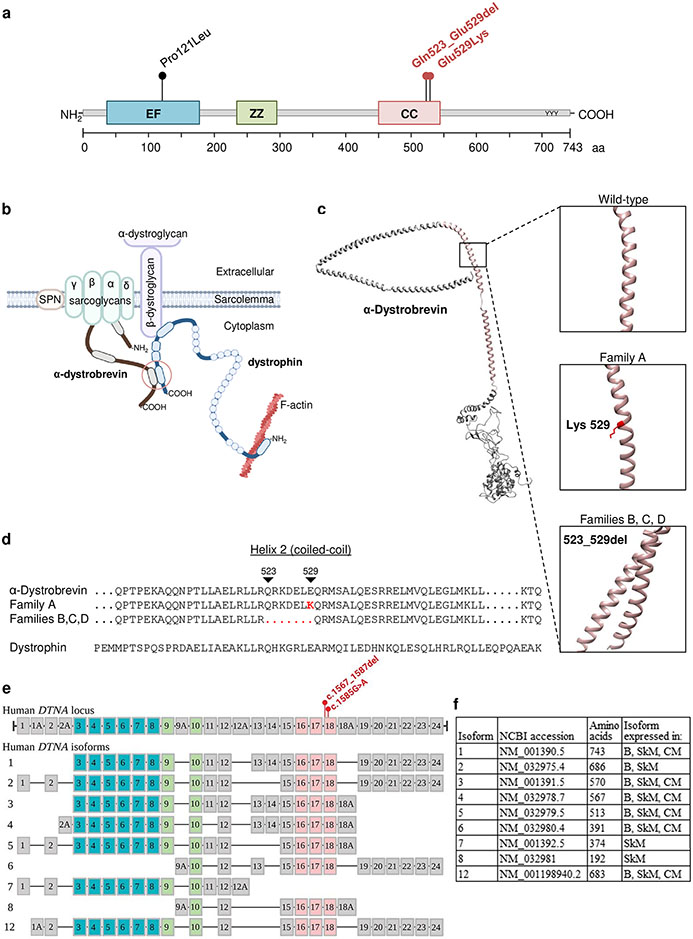
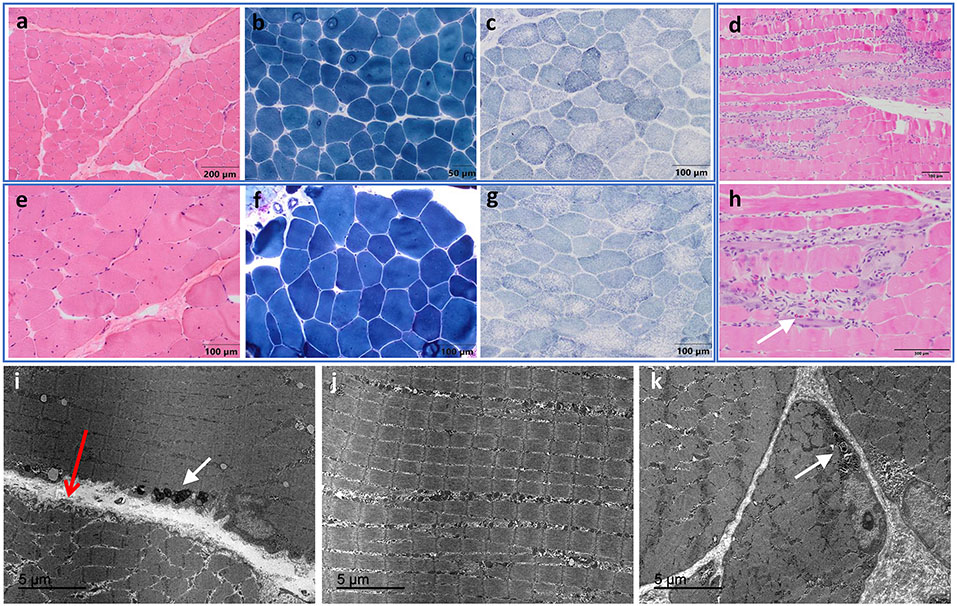
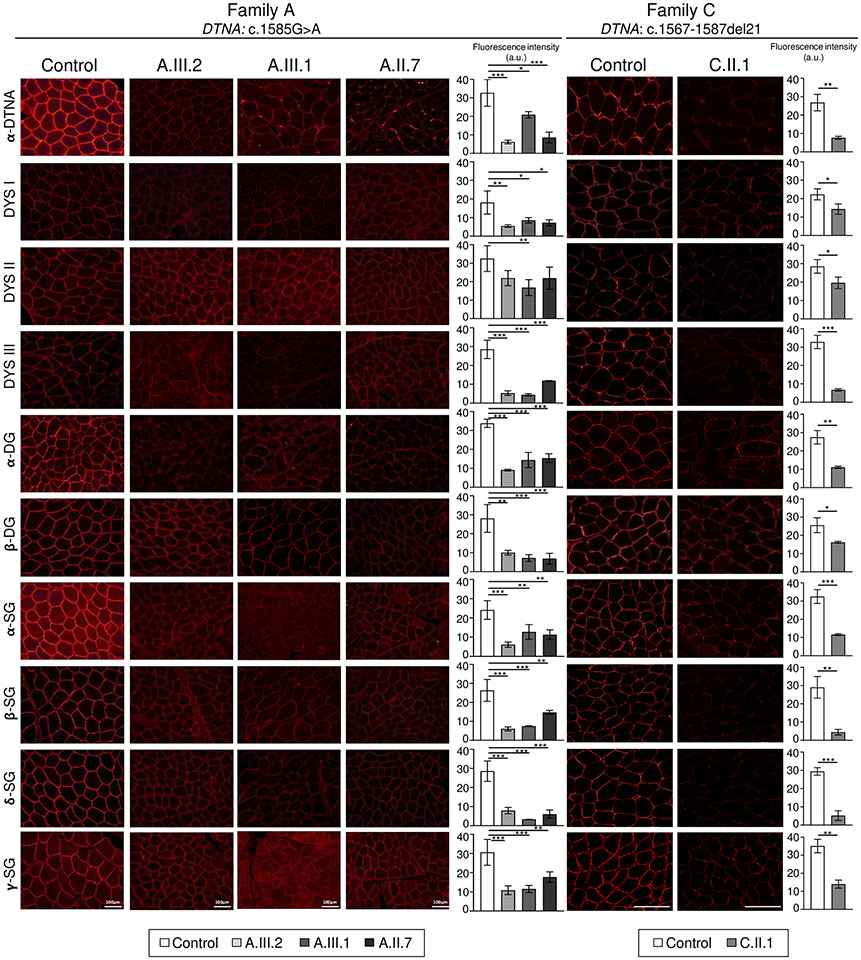
DTNA encodes α-dystrobrevin, a component of the macromolecular dystrophin-glycoprotein complex (DGC) that binds to dystrophin/utrophin and α-syntrophin. Mice lacking α-dystrobrevin have a muscular dystrophy phenotype, but variants in DTNA have not previously been associated with human skeletal muscle disease. We present 12 individuals from four unrelated families with two different monoallelic DTNA variants affecting the coiled-coil domain of α-dystrobrevin. The five affected individuals from family A harbor a c.1585G > A; p.Glu529Lys variant, while the recurrent c.1567_1587del; p.Gln523_Glu529del DTNA variant was identified in the other three families (family B: four affected individuals, family C: one affected individual, and family D: two affected individuals). Myalgia and exercise intolerance, with variable ages of onset, were reported in 10 of 12 affected individuals. Proximal lower limb weakness with onset in the first decade of life was noted in three individuals. Persistent elevations of serum creatine kinase (CK) levels were detected in 11 of 12 affected individuals, 1 of whom had an episode of rhabdomyolysis at 20 years of age. Autism spectrum disorder or learning disabilities were reported in four individuals with the c.1567_1587 deletion. Muscle biopsies in eight affected individuals showed mixed myopathic and dystrophic findings, characterized by fiber size variability, internalized nuclei, and slightly increased extracellular connective tissue and inflammation. Immunofluorescence analysis of biopsies from five affected individuals showed reduced α-dystrobrevin immunoreactivity and variably reduced immunoreactivity of other DGC proteins: dystrophin, α, β, δ and γ-sarcoglycans, and α and β-dystroglycans. The DTNA deletion disrupted an interaction between α-dystrobrevin and syntrophin. Specific variants in the coiled-coil domain of DTNA cause skeletal muscle disease with variable penetrance. Affected individuals show a spectrum of clinical manifestations, with severity ranging from hyperCKemia, myalgias, and exercise intolerance to childhood-onset proximal muscle weakness. Our findings expand the molecular etiologies of both muscular dystrophy and paucisymptomatic hyperCKemia, to now include monoallelic DTNA variants as a novel cause of skeletal muscle disease in humans.
Keywords: Dystrophin; Muscle sarcolemmal proteins; Myalgia; Rhabdomyolysis; hyperCKemia; α-Dystrobrevin.
© 2023. The Author(s), under exclusive licence to Springer-Verlag GmbH Germany, part of Springer Nature.
Conflict of interest statement
Figures



References
-
- Amici DR, Pinal-Fernandez I, Mázala DAG, Lloyd TE, Corse AM, Christopher-Stine L, Mammen AL, Chin ER (2017) Calcium dysregulation, functional calpainopathy, and endoplasmic reticulum stress in sporadic inclusion body myositis. Acta neuropathologica communications 5:24. doi: 10.1186/s40478-017-0427-7 - DOI - PMC - PubMed
-
- Bruels CC, Li C, Mendoza T, Khan J, Reddy HM, Estrella EA, Ghosh PS, Darras BT, Lidov HGW, Pacak CA, Kunkel LM, Modave F, Draper I, Kang PB (2019) Identification of a pathogenic mutation in ATP2A1 via in silico analysis of exome data for cryptic aberrant splice sites. Molecular Genetics and Genomic Medicine 7:e552. doi: 10.1002/mgg3.552 - DOI - PMC - PubMed
-
- Cagliani R, Comi GP, Tancredi L, Sironi M, Fortunate F, Giorda R, Bardoni A, Moggio M, Prelle A, Bresolin N, Scarlato G (2001) Primary beta-sarcoglycanopathy manifesting as recurrent exercise-induced myoglobinuria. Neuromuscular Disorders 11:389–394. doi: 10.1016/S0960-8966(00)00207-8 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
