

CXCL8/CXCR2 signaling mediates bone marrow fibrosis and is a therapeutic target in myelofibrosis
- PMID: 36800567
- PMCID: PMC10273167
- DOI: 10.1182/blood.2022015418
CXCL8/CXCR2 signaling mediates bone marrow fibrosis and is a therapeutic target in myelofibrosis
Abstract
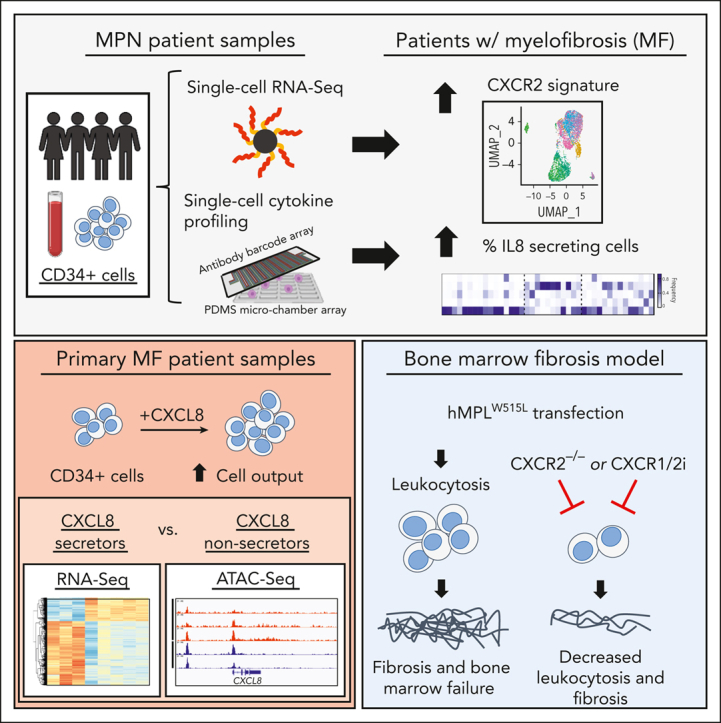
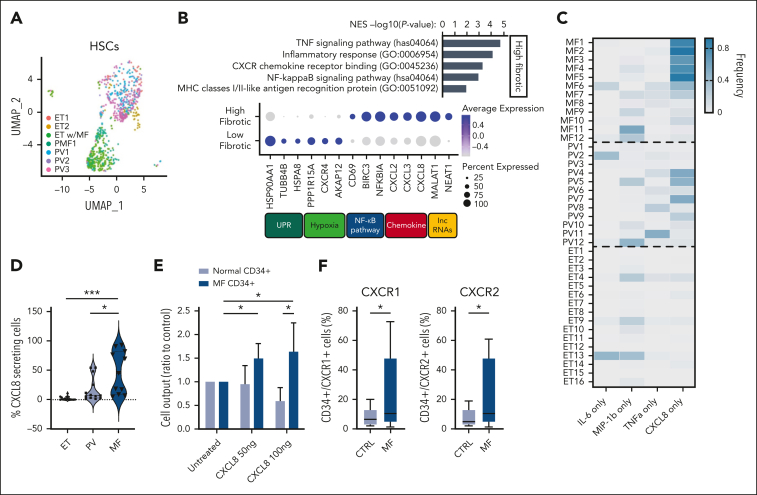
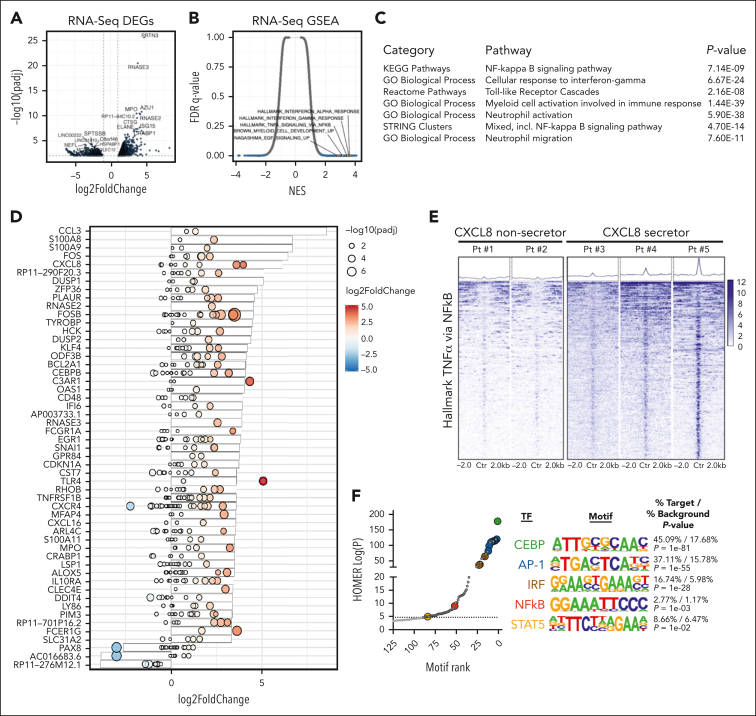
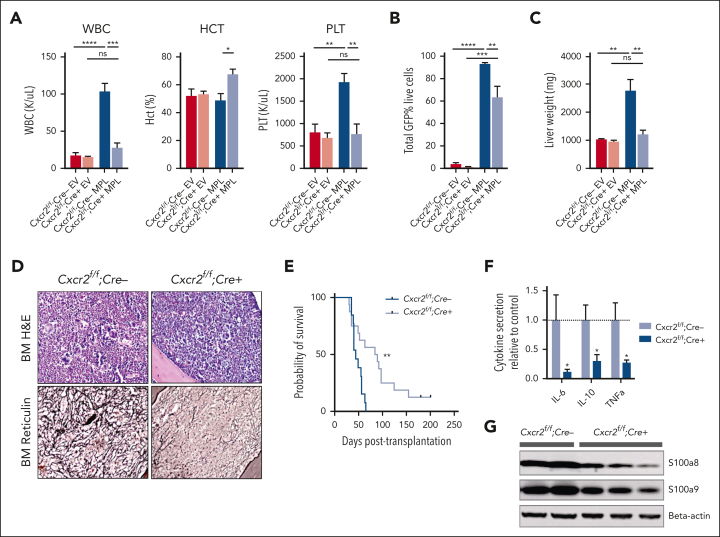
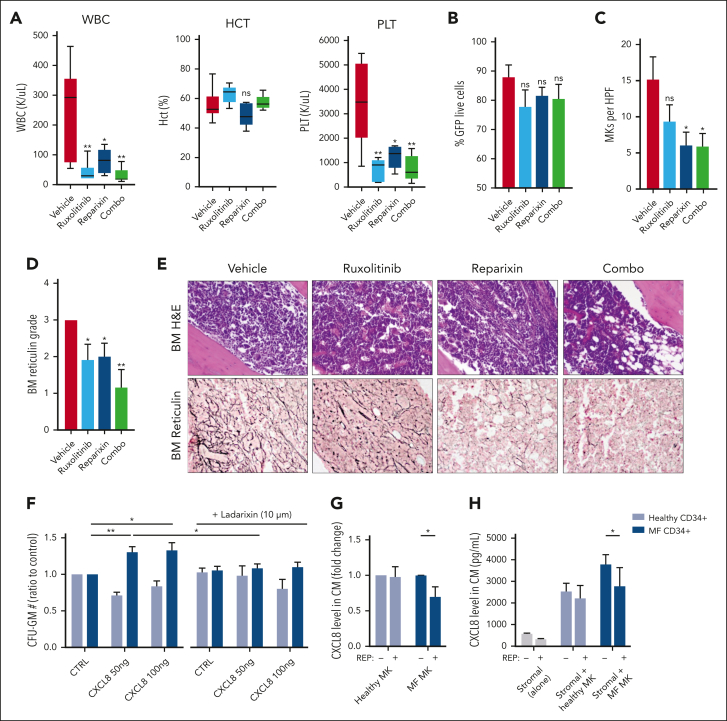
Proinflammatory signaling is a hallmark feature of human cancer, including in myeloproliferative neoplasms (MPNs), most notably myelofibrosis (MF). Dysregulated inflammatory signaling contributes to fibrotic progression in MF; however, the individual cytokine mediators elicited by malignant MPN cells to promote collagen-producing fibrosis and disease evolution are yet to be fully elucidated. Previously, we identified a critical role for combined constitutive JAK/STAT and aberrant NF-κB proinflammatory signaling in MF development. Using single-cell transcriptional and cytokine-secretion studies of primary cells from patients with MF and the human MPLW515L (hMPLW515L) murine model of MF, we extend our previous work and delineate the role of CXCL8/CXCR2 signaling in MF pathogenesis and bone marrow fibrosis progression. Hematopoietic stem/progenitor cells from patients with MF are enriched for a CXCL8/CXCR2 gene signature and display enhanced proliferation and fitness in response to an exogenous CXCL8 ligand in vitro. Genetic deletion of Cxcr2 in the hMPLW515L-adoptive transfer model abrogates fibrosis and extends overall survival, and pharmacologic inhibition of the CXCR1/2 pathway improves hematologic parameters, attenuates bone marrow fibrosis, and synergizes with JAK inhibitor therapy. Our mechanistic insights provide a rationale for therapeutic targeting of the CXCL8/CXCR2 pathway among patients with MF.
© 2023 by The American Society of Hematology. Licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0), permitting only noncommercial, nonderivative use with attribution. All other rights reserved.
Conflict of interest statement
Conflict-of-interest disclosure: R.L.L. is on the supervisory board of Qiagen and is a scientific adviser to Imago, Mission Bio, Bakx, Zentalis, Ajax, Auron, Prelude, C4 Therapeutics, and IsoPlexis; has received research support from AbbVie, Constellation, Ajax, Zentalis, and Prelude; has received research support from and consulted for Celgene and Roche and has consulted for Syndax, Incyte, Janssen, Astellas, MorphoSys, and Novartis; and has received honoraria from AstraZeneca and Novartis for invited lectures and from Gilead and Novartis for grant reviews. A.J.D. has served on an advisory committee for Incyte. R.F. is cofounder and scientific adviser of IsoPlexis, Singleron Biotechnologies, and AtlasXomics with significant financial interest. A.R.M. and R.H. received funds from Dompé farmaceutici S.p.A. (Via Campo di Pile, 67100 L’Aquila, Italy). J.C., E.T., T.M., and J.Z. are employees and equity partners of IsoPlexis Corporation. M.K. is currently an employee of Imago BioSciences. R.L.B. has received honoraria from Mission Bio and is a member of the Speakers Bureau for Mission Bio. The remaining authors declare no competing financial interests.
Figures
Comment in
-
Shelter from the cytokine storm in myelofibrosis.Blood. 2023 May 18;141(20):2415-2416. doi: 10.1182/blood.2023019779. Blood. 2023. PMID: 37200061 No abstract available.
References
-
- Tefferi A, Pardanani A. Myeloproliferative neoplasms: a contemporary review. JAMA Oncol. 2015;1(1):97–105. - PubMed
-
- Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–1061. - PubMed
-
- Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–1790. - PubMed
-
- Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–397. - PubMed
-
- James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–1148. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
