

Clinical feature difference between juvenile amyotrophic lateral sclerosis with SPTLC1 and FUS mutations
- PMID: 36801857
- PMCID: PMC10106144
- DOI: 10.1097/CM9.0000000000002495
Clinical feature difference between juvenile amyotrophic lateral sclerosis with SPTLC1 and FUS mutations
Abstract
Background: Juvenile amyotrophic lateral sclerosis (JALS) is an uncommon form of amyotrophic lateral sclerosis whose age at onset (AAO) is defined as prior to 25 years. FUS mutations are the most common cause of JALS. SPTLC1 was recently identified as a disease-causative gene for JALS, which has rarely been reported in Asian populations. Little is known regarding the difference in clinical features between JALS patients carrying FUS and SPTLC1 mutations. This study aimed to screen mutations in JALS patients and to compare the clinical features between JALS patients with FUS and SPTLC1 mutations.
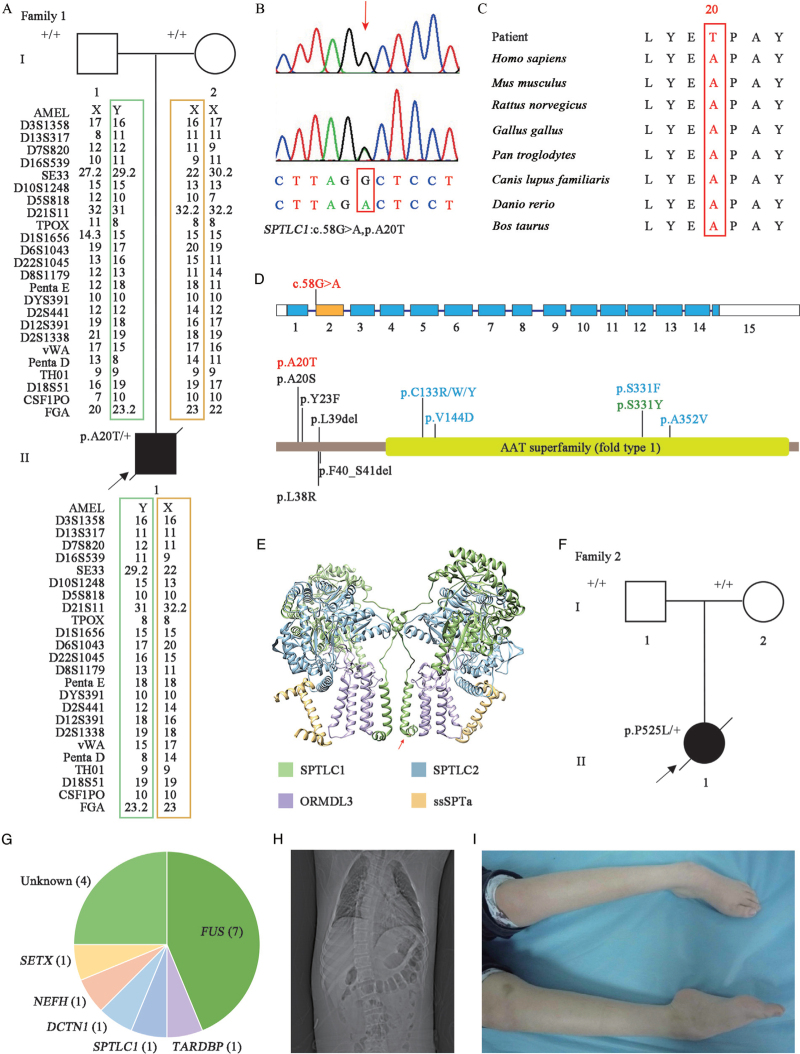
Methods: Sixteen JALS patients were enrolled, including three newly recruited patients between July 2015 and August 2018 from the Second Affiliated Hospital, Zhejiang University School of Medicine. Mutations were screened by whole-exome sequencing. In addition, clinical features such as AAO, onset site and disease duration were extracted and compared between JALS patients carrying FUS and SPTLC1 mutations through a literature review.
Results: A novel and de novo SPTLC1 mutation (c.58G>A, p.A20T) was identified in a sporadic patient. Among 16 JALS patients, 7/16 carried FUS mutations and 5/16 carried respective SPTLC1 , SETX , NEFH , DCTN1 , and TARDBP mutations. Compared with FUS mutation patients, those with SPTLC1 mutations had an earlier AAO (7.9 ± 4.6 years vs. 18.1 ± 3.9 years, P < 0.01), much longer disease duration (512.0 [416.7-607.3] months vs. 33.4 [21.6-45.1] months, P < 0.01), and no onset of bulbar.
Conclusion: Our findings expand the genetic and phenotypic spectrum of JALS and help to better understand the genotype-phenotype correlation of JALS.
Copyright © 2023 The Chinese Medical Association, produced by Wolters Kluwer, Inc. under the CC-BY-NC-ND license.
Conflict of interest statement
None.
Figures
Similar articles
-
The investigation of genetic and clinical features in Chinese patients with juvenile amyotrophic lateral sclerosis.Clin Genet. 2017 Sep;92(3):267-273. doi: 10.1111/cge.13015. Epub 2017 Apr 20. Clin Genet. 2017. PMID: 28429524
-
FUS mutation is probably the most common pathogenic gene for JALS, especially sporadic JALS.Rev Neurol (Paris). 2021 Apr;177(4):333-340. doi: 10.1016/j.neurol.2020.06.010. Epub 2020 Oct 6. Rev Neurol (Paris). 2021. PMID: 33036763 Review.
-
A de novo c.113 T > C: p.L38R mutation of SPTLC1: case report of a girl with sporadic juvenile amyotrophic lateral sclerosis.Amyotroph Lateral Scler Frontotemporal Degener. 2022 Nov;23(7-8):634-637. doi: 10.1080/21678421.2022.2096409. Epub 2022 Oct 7. Amyotroph Lateral Scler Frontotemporal Degener. 2022. PMID: 36204986
-
Novel FUS deletion in a patient with juvenile amyotrophic lateral sclerosis.Arch Neurol. 2012 May;69(5):653-6. doi: 10.1001/archneurol.2011.2499. Arch Neurol. 2012. PMID: 22248478
-
The Occurrence of FUS Mutations in Pediatric Amyotrophic Lateral Sclerosis: A Case Report and Review of the Literature.J Child Neurol. 2020 Jul;35(8):556-562. doi: 10.1177/0883073820915099. Epub 2020 Apr 13. J Child Neurol. 2020. PMID: 32281455 Review.
Cited by
-
Genetic and functional analyses of SPTLC1 in juvenile amyotrophic lateral sclerosis.J Neurol. 2024 Dec 12;272(1):36. doi: 10.1007/s00415-024-12776-5. J Neurol. 2024. PMID: 39666121 Free PMC article.
-
Correlation of single-fiber electromyography studies and functional status in patients with amyotrophic lateral sclerosis.Open Med (Wars). 2024 Jun 29;19(1):20240990. doi: 10.1515/med-2024-0990. eCollection 2024. Open Med (Wars). 2024. PMID: 38953009 Free PMC article.
References
-
- van Es MA, Hardiman O, Chio A, Al-Chalabi A, Pasterkamp RJ, Veldink JH, et al. . Amyotrophic lateral sclerosis. Lancet 2017; 390:2084–2098. doi: 10.1016/S0140-6736(17)31287-4. - PubMed
-
- Orban P, Devon RS, Hayden MR, Leavitt BR. Chapter 15 juvenile amyotrophic lateral sclerosis. Handb Clin Neurol 2007; 82:301–312. doi: 10.1016/S0072-9752(07)80018-2. - PubMed
-
- Dawkins JL, Hulme DJ, Brahmbhatt SB, Auer-Grumbach M, Nicholson GA. Mutations in SPTLC1, encoding serine palmitoyltransferase, long chain base subunit-1, cause hereditary sensory neuropathy type I. Nat Genet 2001; 27:309–312. doi: 10.1038/85879. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
