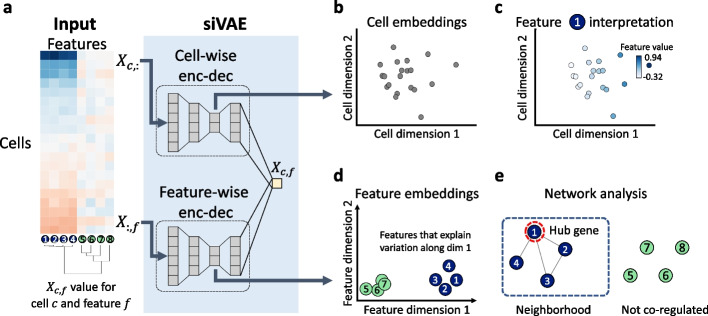
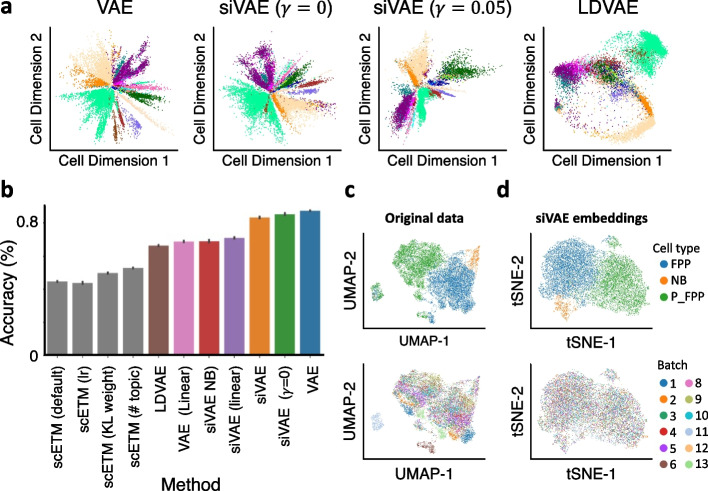
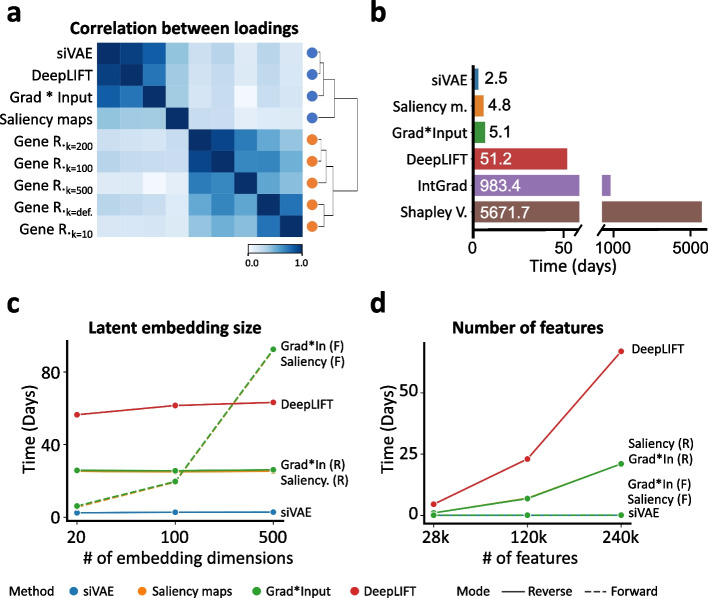
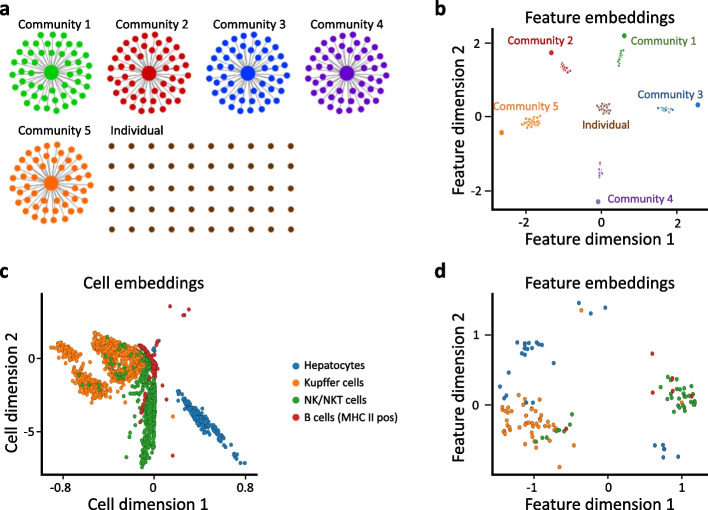
siVAE: interpretable deep generative models for single-cell transcriptomes
- PMID: 36803416
- PMCID: PMC9940350
- DOI: 10.1186/s13059-023-02850-y
siVAE: interpretable deep generative models for single-cell transcriptomes
Abstract
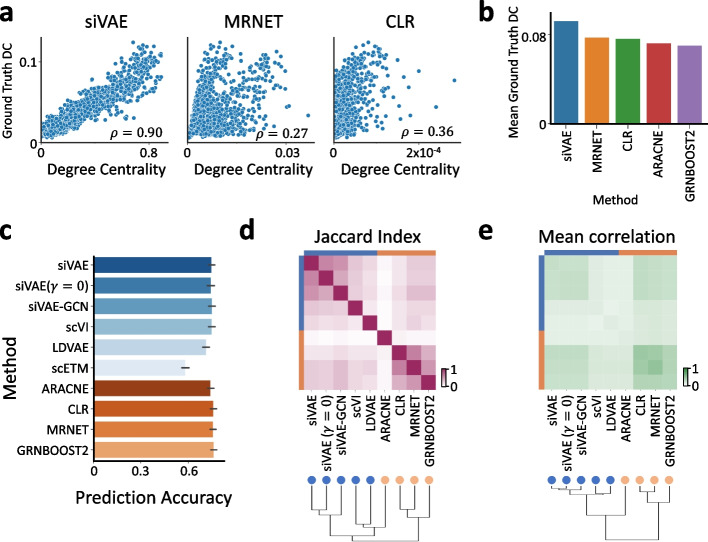
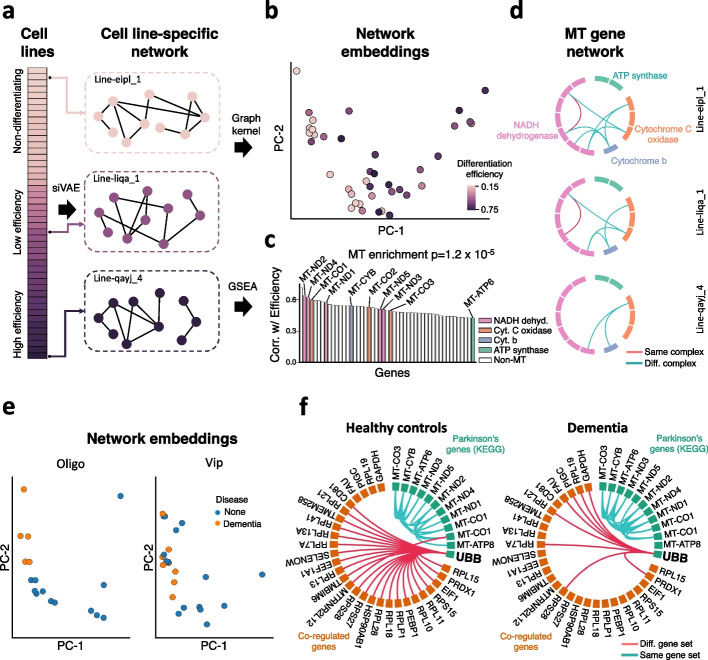
Neural networks such as variational autoencoders (VAE) perform dimensionality reduction for the visualization and analysis of genomic data, but are limited in their interpretability: it is unknown which data features are represented by each embedding dimension. We present siVAE, a VAE that is interpretable by design, thereby enhancing downstream analysis tasks. Through interpretation, siVAE also identifies gene modules and hubs without explicit gene network inference. We use siVAE to identify gene modules whose connectivity is associated with diverse phenotypes such as iPSC neuronal differentiation efficiency and dementia, showcasing the wide applicability of interpretable generative models for genomic data analysis.
© 2023. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures