

The landscape of antibody binding affinity in SARS-CoV-2 Omicron BA.1 evolution
- PMID: 36803543
- PMCID: PMC9949795
- DOI: 10.7554/eLife.83442
The landscape of antibody binding affinity in SARS-CoV-2 Omicron BA.1 evolution
Abstract
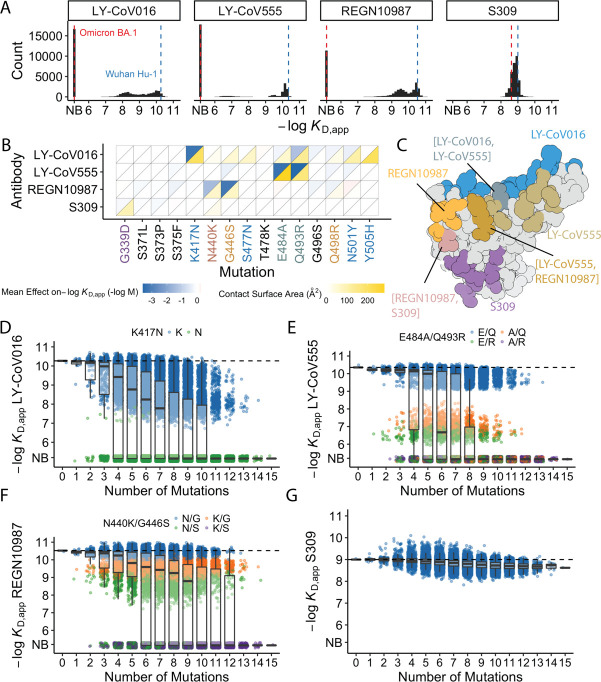
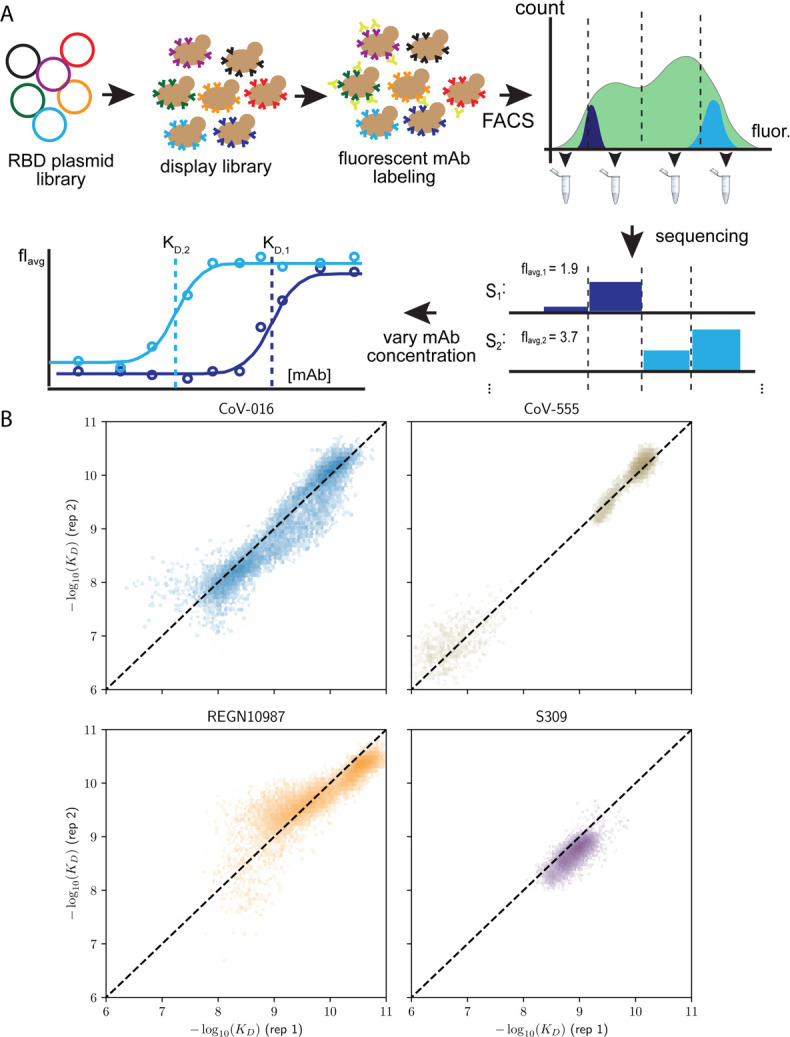
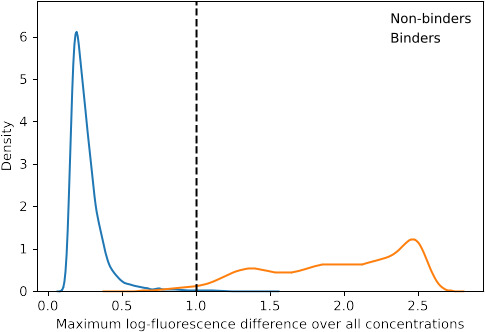
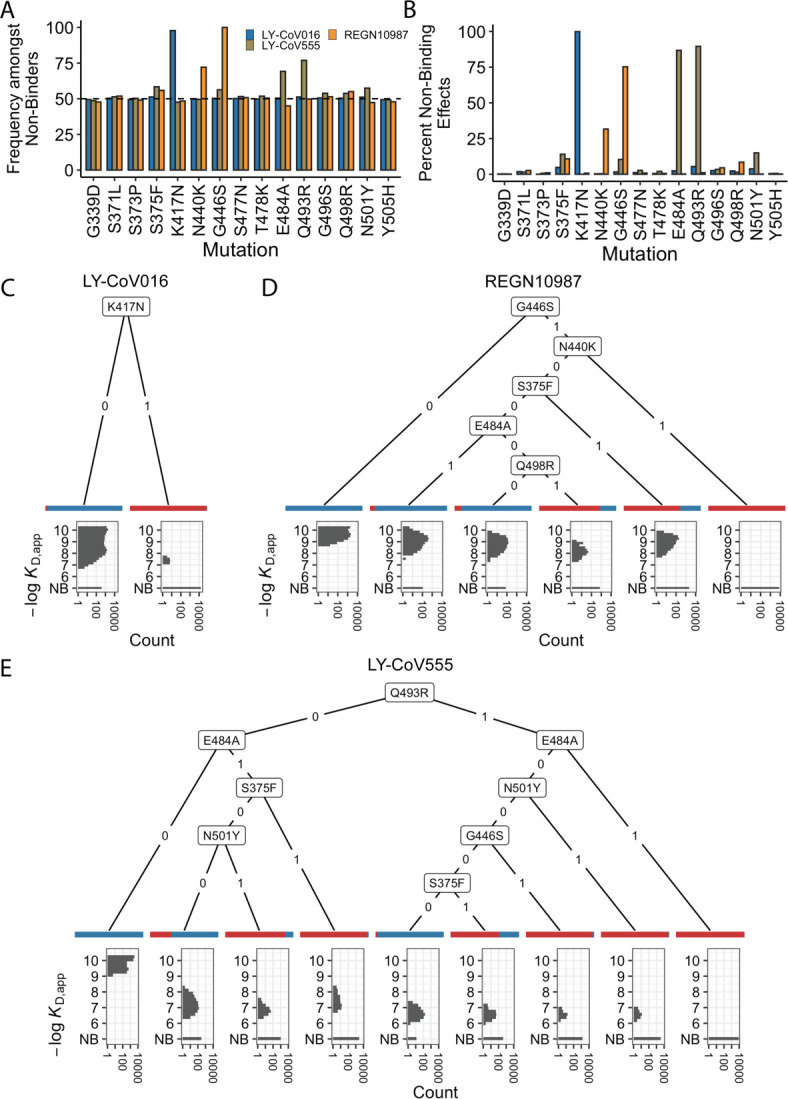
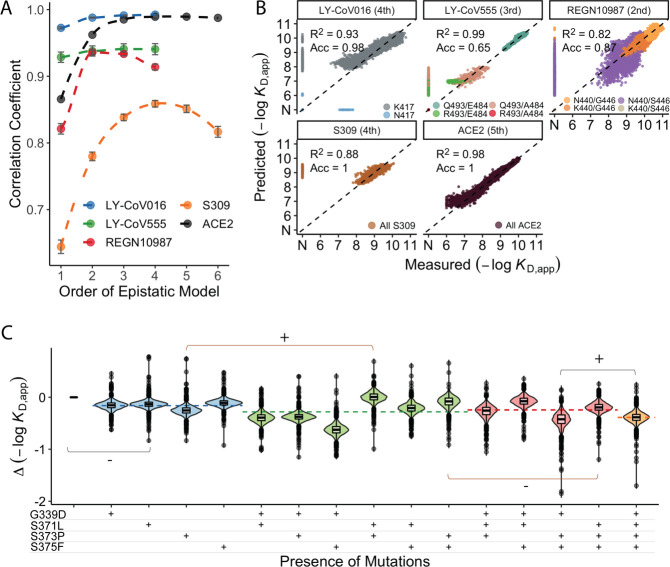
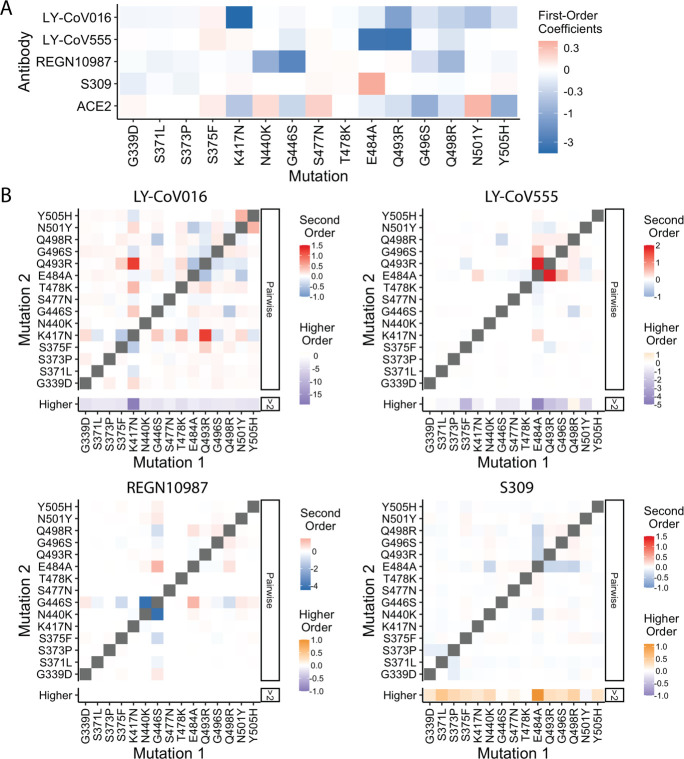
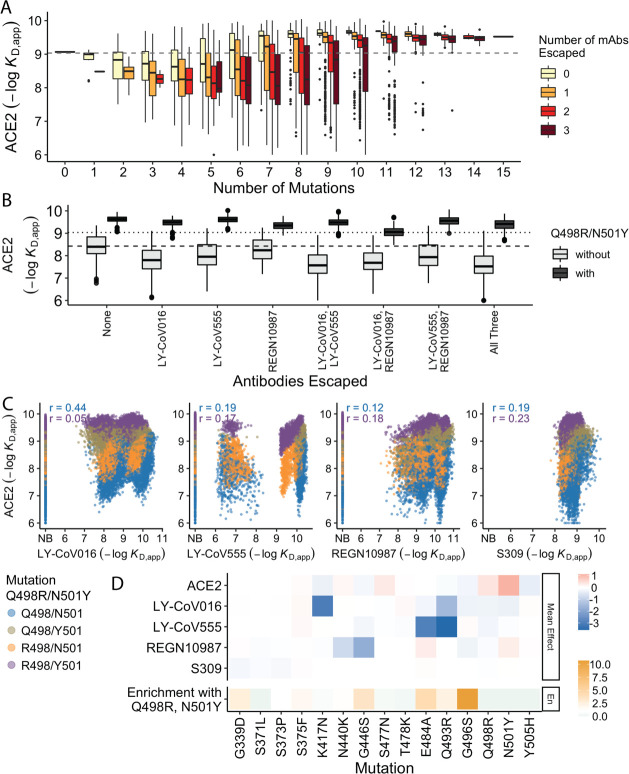
The Omicron BA.1 variant of SARS-CoV-2 escapes convalescent sera and monoclonal antibodies that are effective against earlier strains of the virus. This immune evasion is largely a consequence of mutations in the BA.1 receptor binding domain (RBD), the major antigenic target of SARS-CoV-2. Previous studies have identified several key RBD mutations leading to escape from most antibodies. However, little is known about how these escape mutations interact with each other and with other mutations in the RBD. Here, we systematically map these interactions by measuring the binding affinity of all possible combinations of these 15 RBD mutations (215=32,768 genotypes) to 4 monoclonal antibodies (LY-CoV016, LY-CoV555, REGN10987, and S309) with distinct epitopes. We find that BA.1 can lose affinity to diverse antibodies by acquiring a few large-effect mutations and can reduce affinity to others through several small-effect mutations. However, our results also reveal alternative pathways to antibody escape that does not include every large-effect mutation. Moreover, epistatic interactions are shown to constrain affinity decline in S309 but only modestly shape the affinity landscapes of other antibodies. Together with previous work on the ACE2 affinity landscape, our results suggest that the escape of each antibody is mediated by distinct groups of mutations, whose deleterious effects on ACE2 affinity are compensated by another distinct group of mutations (most notably Q498R and N501Y).
Keywords: S. cerevisiae; SARS-CoV-2; antibody escape; evolutionary biology; fitness landscape; immune evasion; omicron variant.
© 2023, Moulana, Dupic et al.
Conflict of interest statement
AM, TD, JC, AR No competing interests declared, AP, MD has or has recently consulted for Leyden Labs, AG, TS is an inventor on Fred Hutch licensed patents related to viral deep mutational scanning (patent numbers WO2022146484, WO2020006494 and application number US20210147832), JB is an inventor on Fred Hutch licensed patents related to viral deep mutational scanning (patent numbers WO2022146484, WO2020006494 and application number US20210147832). JDB has or has recently consulted for Apriori Bio, Oncorus, Moderna, and Merck
Figures
References
-
- Barnes CO, Jette CA, Abernathy ME, Dam KMA, Esswein SR, Gristick HB, Malyutin AG, Sharaf NG, Huey-Tubman KE, Lee YE, Robbiani DF, Nussenzweig MC, West AP, Bjorkman PJ. SARS-cov-2 neutralizing antibody structures inform therapeutic strategies. Nature. 2020a;588:682–687. doi: 10.1038/s41586-020-2852-1. - DOI - PMC - PubMed
-
- Barnes CO, West AP, Huey-Tubman KE, Hoffmann MAG, Sharaf NG, Hoffman PR, Koranda N, Gristick HB, Gaebler C, Muecksch F, Lorenzi JCC, Finkin S, Hägglöf T, Hurley A, Millard KG, Weisblum Y, Schmidt F, Hatziioannou T, Bieniasz PD, Caskey M, Robbiani DF, Nussenzweig MC, Bjorkman PJ. Structures of human antibodies bound to SARS-cov-2 spike reveal common epitopes and recurrent features of antibodies. Cell. 2020b;182:828–842. doi: 10.1016/j.cell.2020.06.025. - DOI - PMC - PubMed
-
- Borkovec M. Ggplot” visualizations for the “partykit” package. 1.0.0Ggplot. 2019 https://cran.r-project.org/web/packages/ggparty/ggparty.pdf
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
