

Comparison of long- and short-read metagenomic assembly for low-abundance species and resistance genes
- PMID: 36804804
- PMCID: PMC10025444
- DOI: 10.1093/bib/bbad050
Comparison of long- and short-read metagenomic assembly for low-abundance species and resistance genes
Abstract
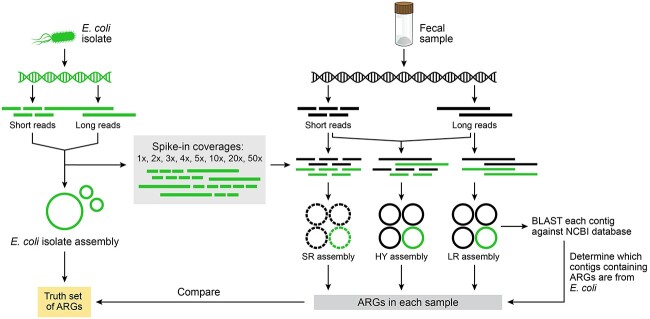
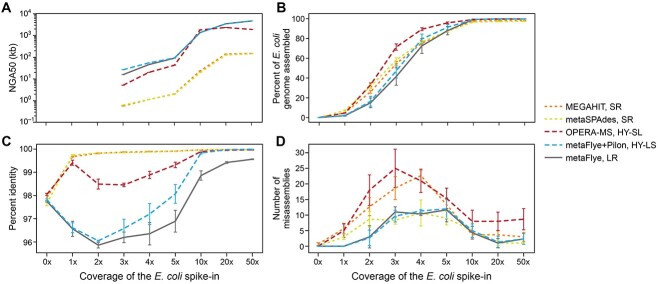
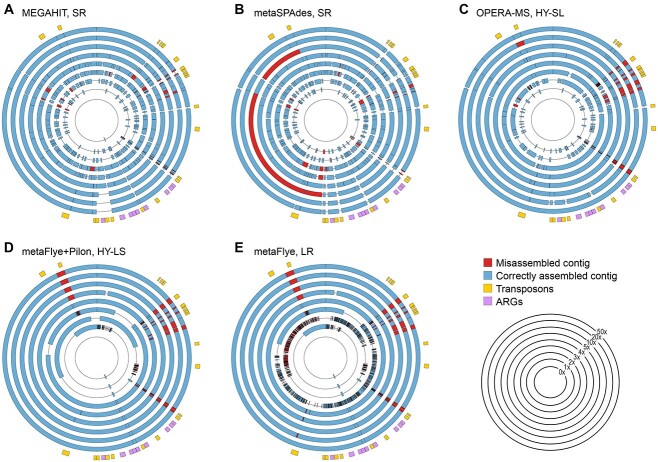
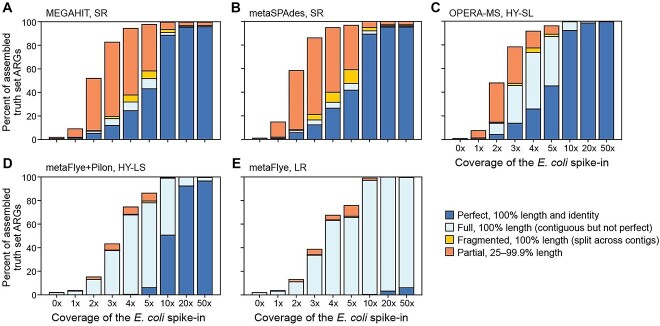
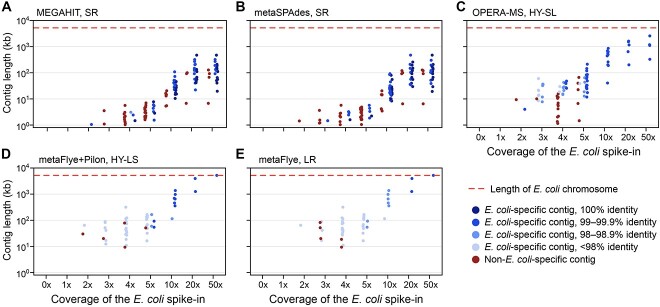
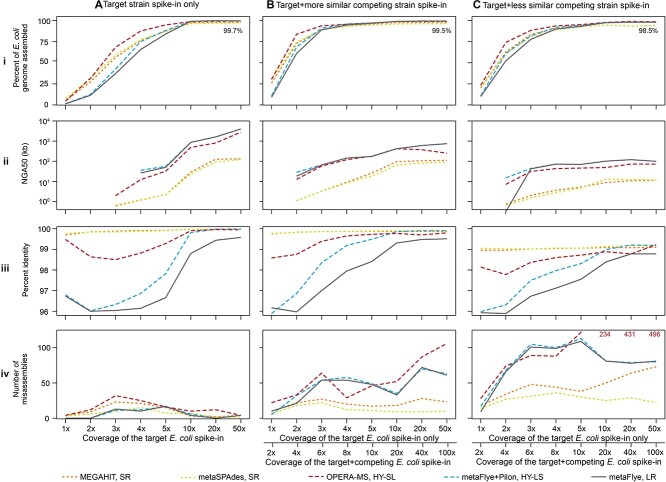
Recent technological and computational advances have made metagenomic assembly a viable approach to achieving high-resolution views of complex microbial communities. In previous benchmarking, short-read (SR) metagenomic assemblers had the highest accuracy, long-read (LR) assemblers generated the most contiguous sequences and hybrid (HY) assemblers balanced length and accuracy. However, no assessments have specifically compared the performance of these assemblers on low-abundance species, which include clinically relevant organisms in the gut. We generated semi-synthetic LR and SR datasets by spiking small and increasing amounts of Escherichia coli isolate reads into fecal metagenomes and, using different assemblers, examined E. coli contigs and the presence of antibiotic resistance genes (ARGs). For ARG assembly, although SR assemblers recovered more ARGs with high accuracy, even at low coverages, LR assemblies allowed for the placement of ARGs within longer, E. coli-specific contigs, thus pinpointing their taxonomic origin. HY assemblies identified resistance genes with high accuracy and had lower contiguity than LR assemblies. Each assembler type's strengths were maintained even when our isolate was spiked in with a competing strain, which fragmented and reduced the accuracy of all assemblies. For strain characterization and determining gene context, LR assembly is optimal, while for base-accurate gene identification, SR assemblers outperform other options. HY assembly offers contiguity and base accuracy, but requires generating data on multiple platforms, and may suffer high misassembly rates when strain diversity exists. Our results highlight the trade-offs associated with each approach for recovering low-abundance taxa, and that the optimal approach is goal-dependent.
Keywords: antibiotic resistance; assembly benchmarking; long reads; low abundance; metagenomic assembly; plasmid assembly.
© The Author(s) 2023. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
Similar articles
-
Metagenomic assemblies tend to break around antibiotic resistance genes.BMC Genomics. 2024 Oct 14;25(1):959. doi: 10.1186/s12864-024-10876-0. BMC Genomics. 2024. PMID: 39402510 Free PMC article.
-
Intestinal microbiota domination under extreme selective pressures characterized by metagenomic read cloud sequencing and assembly.BMC Bioinformatics. 2019 Dec 2;20(Suppl 16):585. doi: 10.1186/s12859-019-3073-1. BMC Bioinformatics. 2019. PMID: 31787070 Free PMC article.
-
Hybrid metagenomic assembly enables high-resolution analysis of resistance determinants and mobile elements in human microbiomes.Nat Biotechnol. 2019 Aug;37(8):937-944. doi: 10.1038/s41587-019-0191-2. Epub 2019 Jul 29. Nat Biotechnol. 2019. PMID: 31359005
-
Assessment of metagenomic assemblers based on hybrid reads of real and simulated metagenomic sequences.Brief Bioinform. 2020 May 21;21(3):777-790. doi: 10.1093/bib/bbz025. Brief Bioinform. 2020. PMID: 30860572 Free PMC article. Review.
-
New approaches for metagenome assembly with short reads.Brief Bioinform. 2020 Mar 23;21(2):584-594. doi: 10.1093/bib/bbz020. Brief Bioinform. 2020. PMID: 30815668 Free PMC article. Review.
Cited by
-
Metagenomic assemblies tend to break around antibiotic resistance genes.BMC Genomics. 2024 Oct 14;25(1):959. doi: 10.1186/s12864-024-10876-0. BMC Genomics. 2024. PMID: 39402510 Free PMC article.
-
Bacterial dynamics of the plastisphere microbiome exposed to sub-lethal antibiotic pollution.Microbiome. 2024 May 24;12(1):97. doi: 10.1186/s40168-024-01803-2. Microbiome. 2024. PMID: 38790062 Free PMC article.
-
Enhancing Clinical Utility: Utilization of International Standards and Guidelines for Metagenomic Sequencing in Infectious Disease Diagnosis.Int J Mol Sci. 2024 Mar 15;25(6):3333. doi: 10.3390/ijms25063333. Int J Mol Sci. 2024. PMID: 38542307 Free PMC article. Review.
-
Ultra pure high molecular weight DNA from soil for Nanopore shotgun metagenomics and metabarcoding sequencing.MethodsX. 2024 Dec 28;14:103134. doi: 10.1016/j.mex.2024.103134. eCollection 2025 Jun. MethodsX. 2024. PMID: 39846015 Free PMC article.
-
ARGContextProfiler: extracting and scoring the genomic contexts of antibiotic resistance genes using assembly graphs.Front Microbiol. 2025 May 21;16:1604461. doi: 10.3389/fmicb.2025.1604461. eCollection 2025. Front Microbiol. 2025. PMID: 40469725 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
