

Platelet-instructed SPP1+ macrophages drive myofibroblast activation in fibrosis in a CXCL4-dependent manner
- PMID: 36807143
- PMCID: PMC9992450
- DOI: 10.1016/j.celrep.2023.112131
Platelet-instructed SPP1+ macrophages drive myofibroblast activation in fibrosis in a CXCL4-dependent manner
Abstract
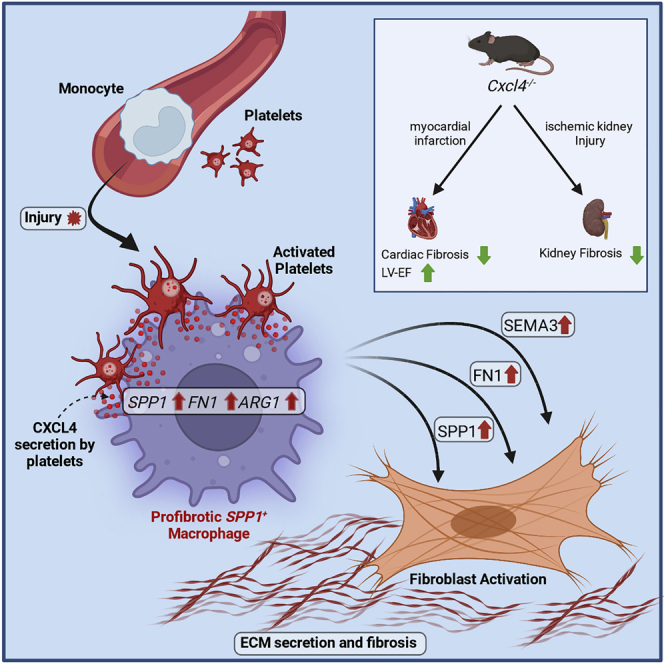
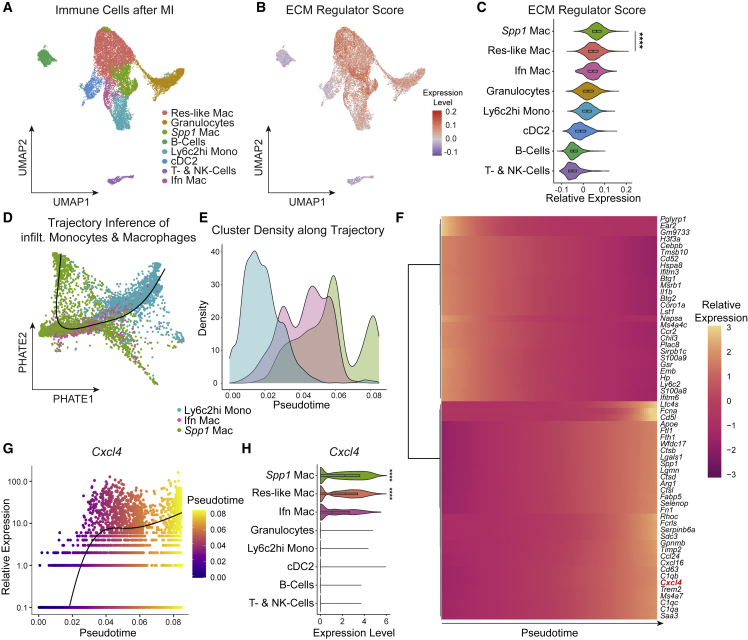
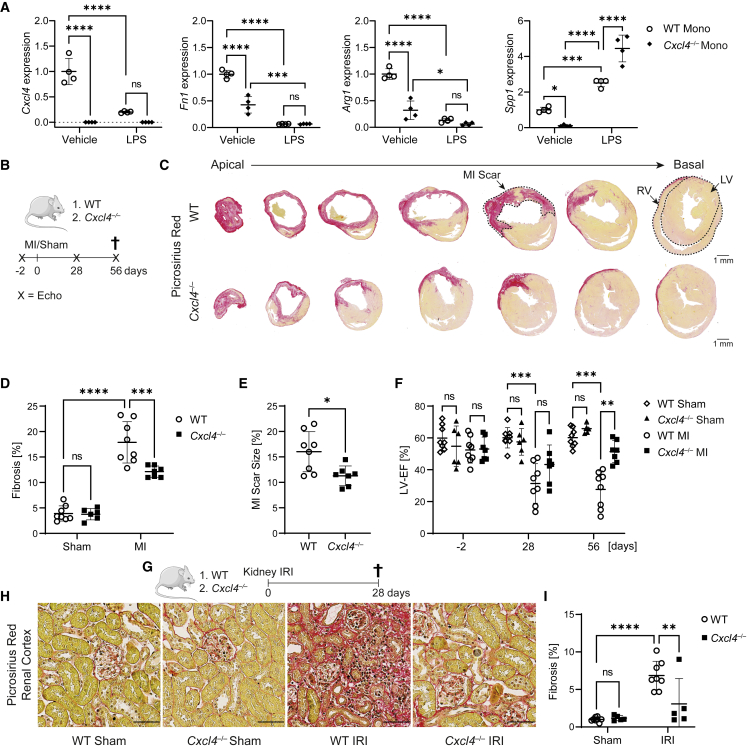
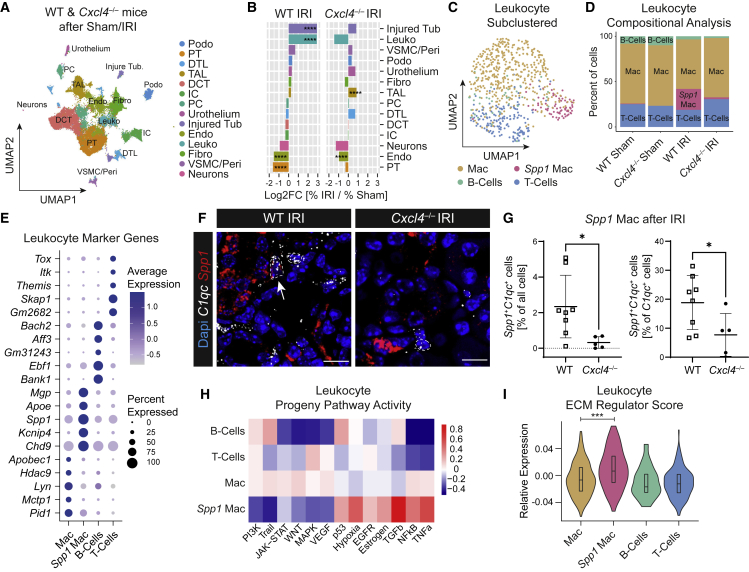
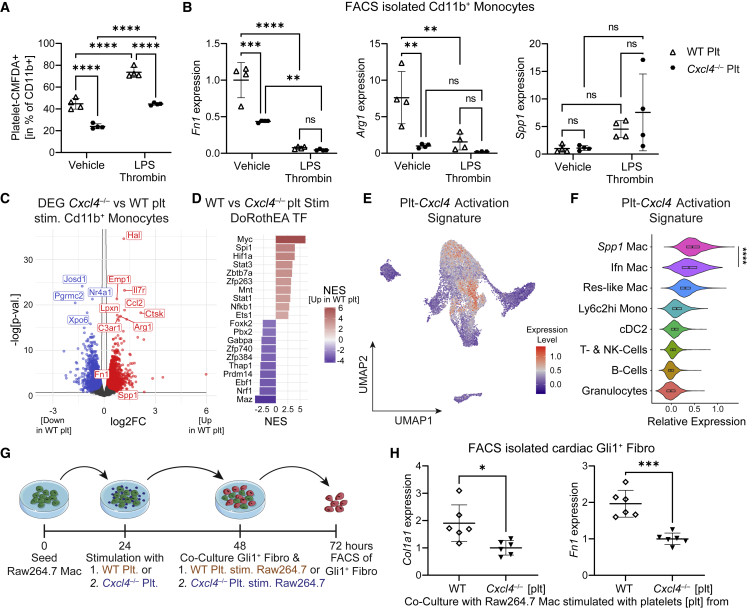
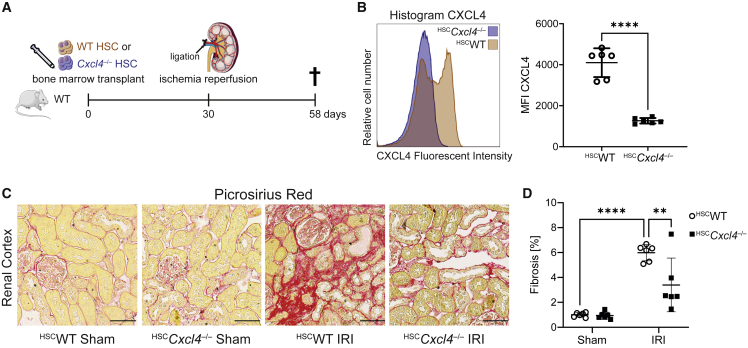
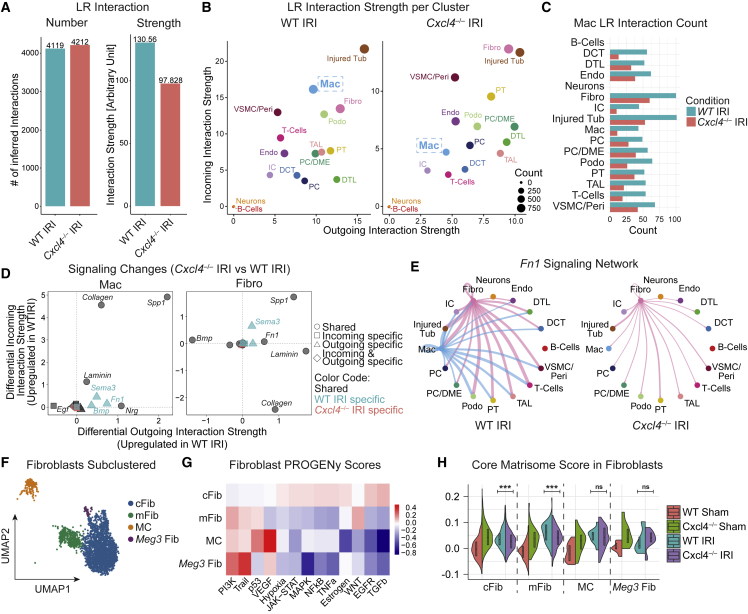
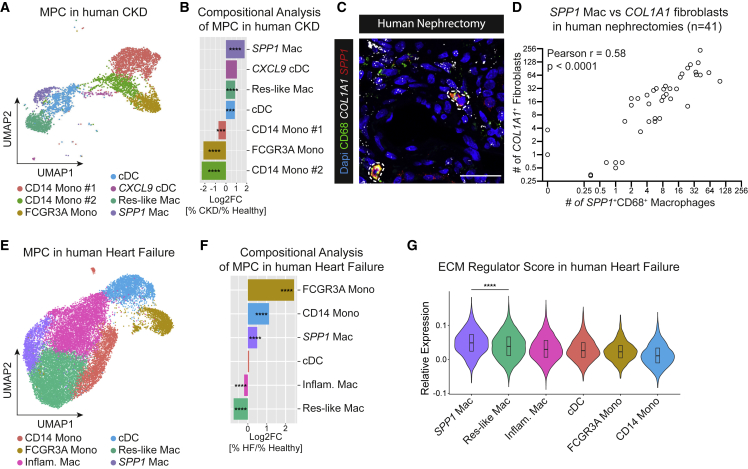
Fibrosis represents the common end stage of chronic organ injury independent of the initial insult, destroying tissue architecture and driving organ failure. Here we discover a population of profibrotic macrophages marked by expression of Spp1, Fn1, and Arg1 (termed Spp1 macrophages), which expands after organ injury. Using an unbiased approach, we identify the chemokine (C-X-C motif) ligand 4 (CXCL4) to be among the top upregulated genes during profibrotic Spp1 macrophage differentiation. In vitro and in vivo studies show that loss of Cxcl4 abrogates profibrotic Spp1 macrophage differentiation and ameliorates fibrosis after both heart and kidney injury. Moreover, we find that platelets, the most abundant source of CXCL4 in vivo, drive profibrotic Spp1 macrophage differentiation. Single nuclear RNA sequencing with ligand-receptor interaction analysis reveals that macrophages orchestrate fibroblast activation via Spp1, Fn1, and Sema3 crosstalk. Finally, we confirm that Spp1 macrophages expand in both human chronic kidney disease and heart failure.
Keywords: CP: Immunology; CXCL4; PF4; SPP1; SPP1 macrophages; chronic kidney disease; fibrosis; heart failure; innate immunity; myocardial infarction; platelets.
Copyright © 2023 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
-
- Kramann R., DiRocco D.P., Humphreys B.D. Understanding the origin, activation and regulation of matrix-producing myofibroblasts for treatment of fibrotic disease. J. Pathol. 2013;231:273–289. - PubMed
-
- Soliman H., Paylor B., Scott R.W., Lemos D.R., Chang C., Arostegui M., Low M., Lee C., Fiore D., Braghetta P., et al. Pathogenic potential of Hic1-expressing cardiac stromal progenitors. Cell Stem Cell. 2020;26:205–220.e8. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
