

ALL-tRNAseq enables robust tRNA profiling in tissue samples
- PMID: 36810209
- PMCID: PMC10111867
- DOI: 10.1101/gad.350233.122
ALL-tRNAseq enables robust tRNA profiling in tissue samples
Abstract
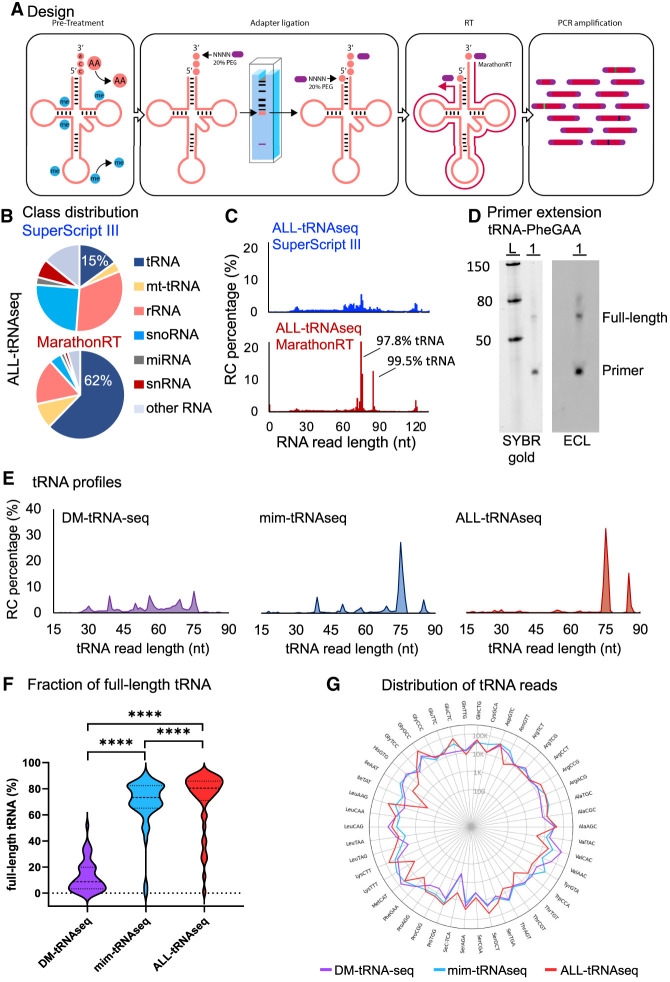
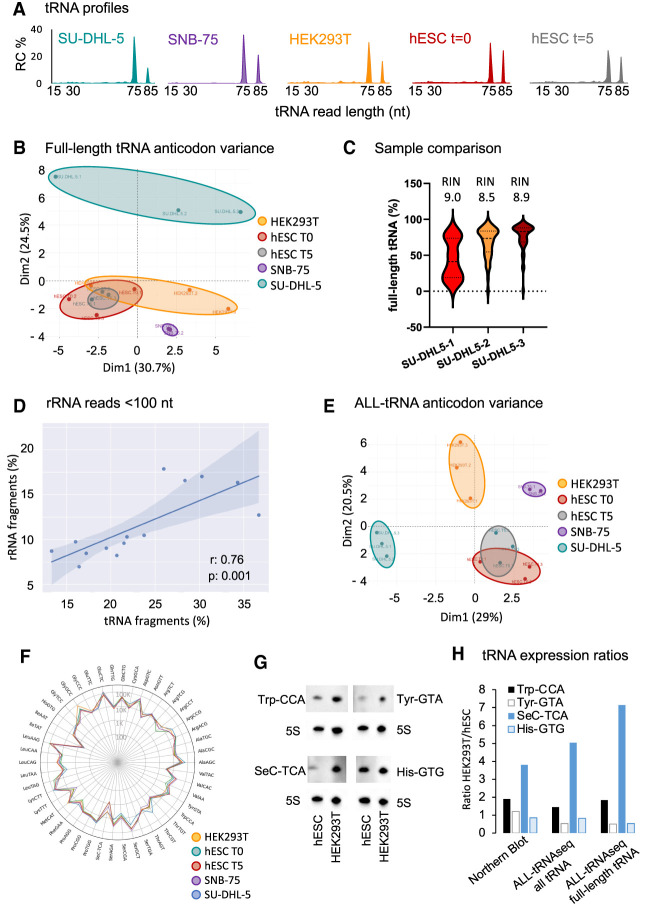
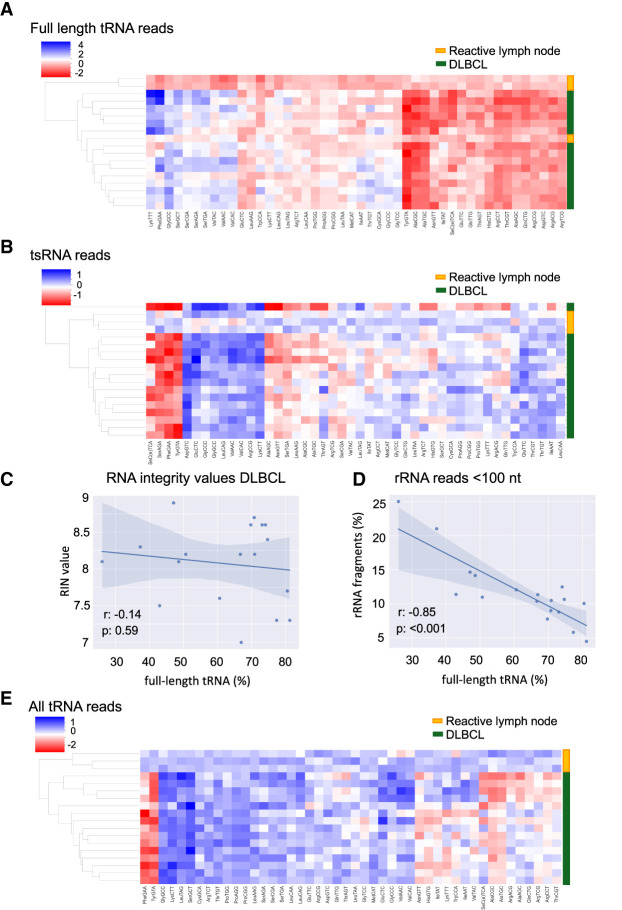
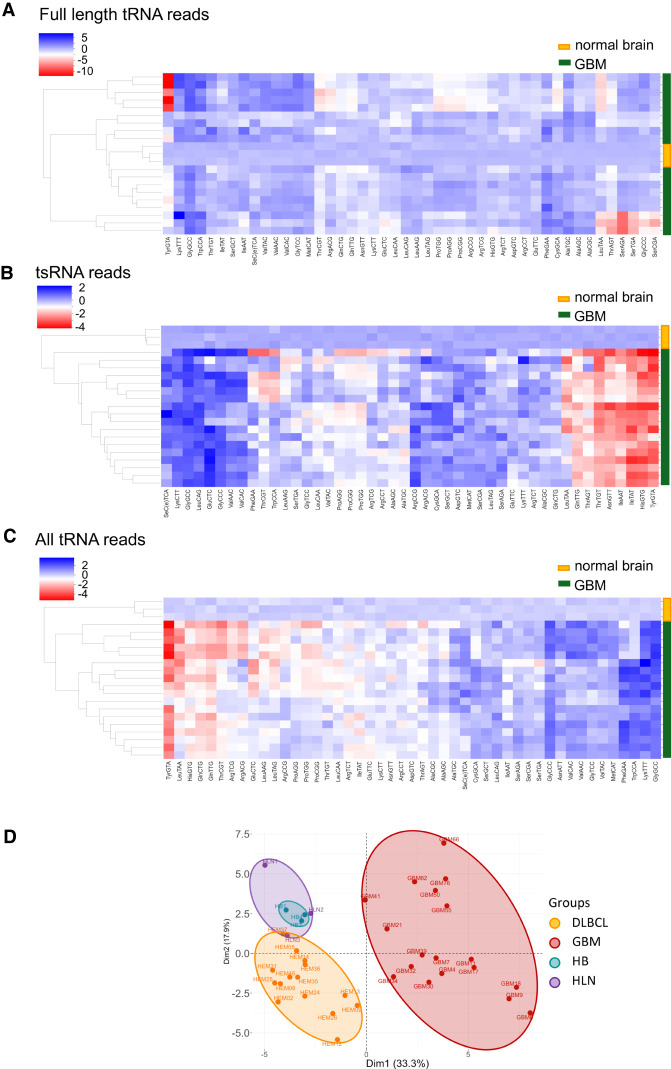
Transfer RNAs (tRNAs) are small adaptor RNAs essential for mRNA translation. Alterations in the cellular tRNA population can directly affect mRNA decoding rates and translational efficiency during cancer development and progression. To evaluate changes in the composition of the tRNA pool, multiple sequencing approaches have been developed to overcome reverse transcription blocks caused by the stable structures of these molecules and their numerous base modifications. However, it remains unclear whether current sequencing protocols faithfully capture tRNAs existing in cells or tissues. This is specifically challenging for clinical tissue samples that often present variable RNA qualities. For this reason, we developed ALL-tRNAseq, which combines the highly processive MarathonRT and RNA demethylation for the robust assessment of tRNA expression, together with a randomized adapter ligation strategy prior to reverse transcription to assess tRNA fragmentation levels in both cell lines and tissues. Incorporation of tRNA fragments not only informed on sample integrity but also significantly improved tRNA profiling of tissue samples. Our data showed that our profiling strategy effectively improves classification of oncogenic signatures in glioblastoma and diffuse large B-cell lymphoma tissues, particularly for samples presenting higher levels of RNA fragmentation, further highlighting the utility of ALL-tRNAseq for translational research.
Keywords: cancer; high-throughput sequencing; tissue samples; transfer RNA.
© 2023 Scheepbouwer et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Aparicio-Puerta E, Lebrón R, Rueda A, Gómez-Martín C, Giannoukakos S, Jaspez D, Medina JM, Zubkovic A, Jurak I, Fromm B, et al. 2019. sRNAbench and sRNAtoolbox 2019: intuitive fast small RNA profiling and differential expression. Nucleic Acids Res 47: W530–W535. 10.1093/nar/gkz415 - DOI - PMC - PubMed
-
- Aparicio-Puerta E, Gómez-Martín C, Giannoukakos S, Medina JM, Scheepbouwer C, García-Moreno A, Carmona-Saez P, Fromm B, Pegtel M, Keller A, et al. 2022. sRNAbench and sRNAtoolbox 2022 update: accurate miRNA and sncRNA profiling for model and non-model organisms. Nucleic Acids Res 50: W710–W717. 10.1093/nar/gkac363 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials