

Elucidation of the GSK3α Structure Informs the Design of Novel, Paralog-Selective Inhibitors
- PMID: 36812145
- PMCID: PMC10020971
- DOI: 10.1021/acschemneuro.2c00476
Elucidation of the GSK3α Structure Informs the Design of Novel, Paralog-Selective Inhibitors
Abstract
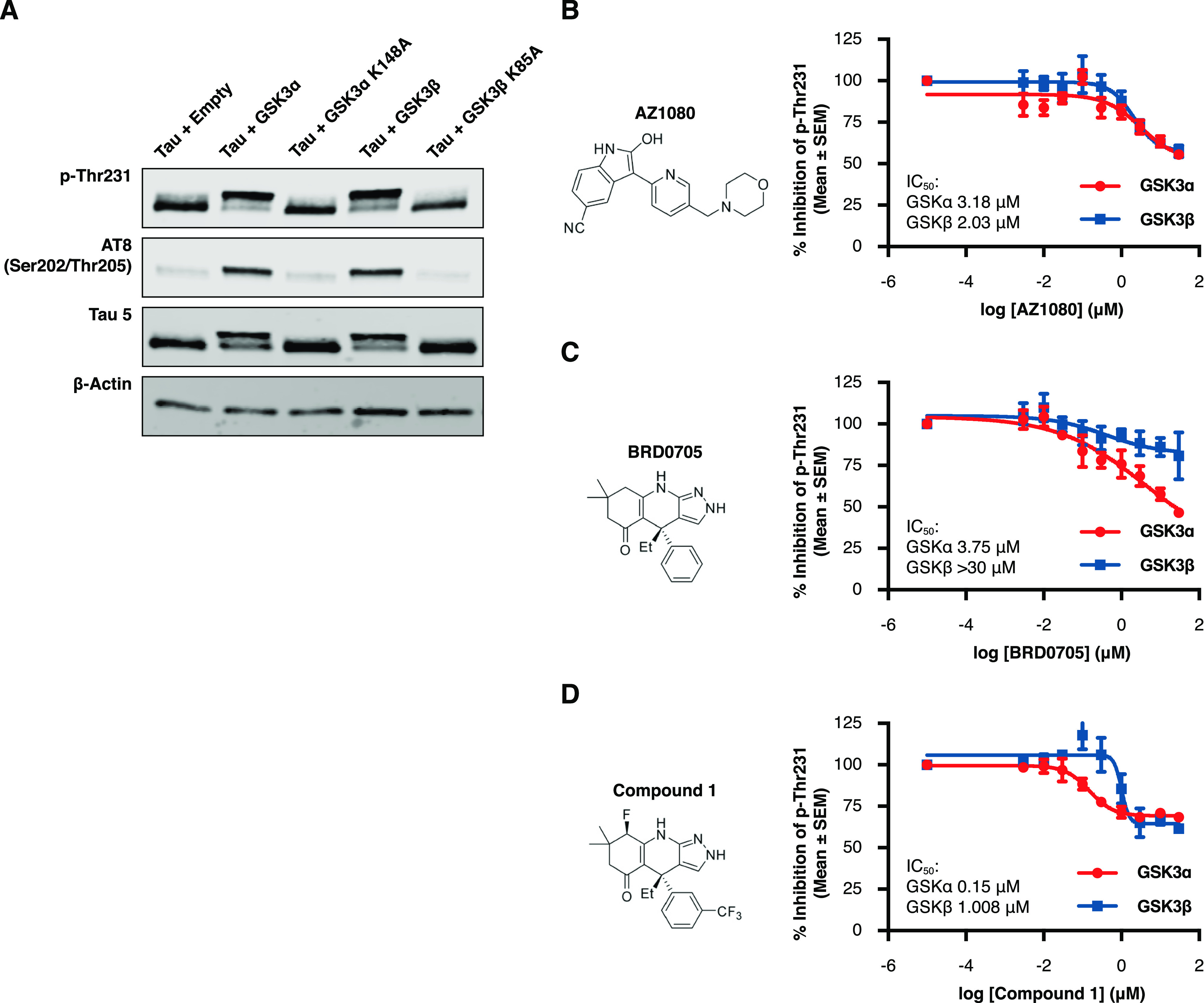
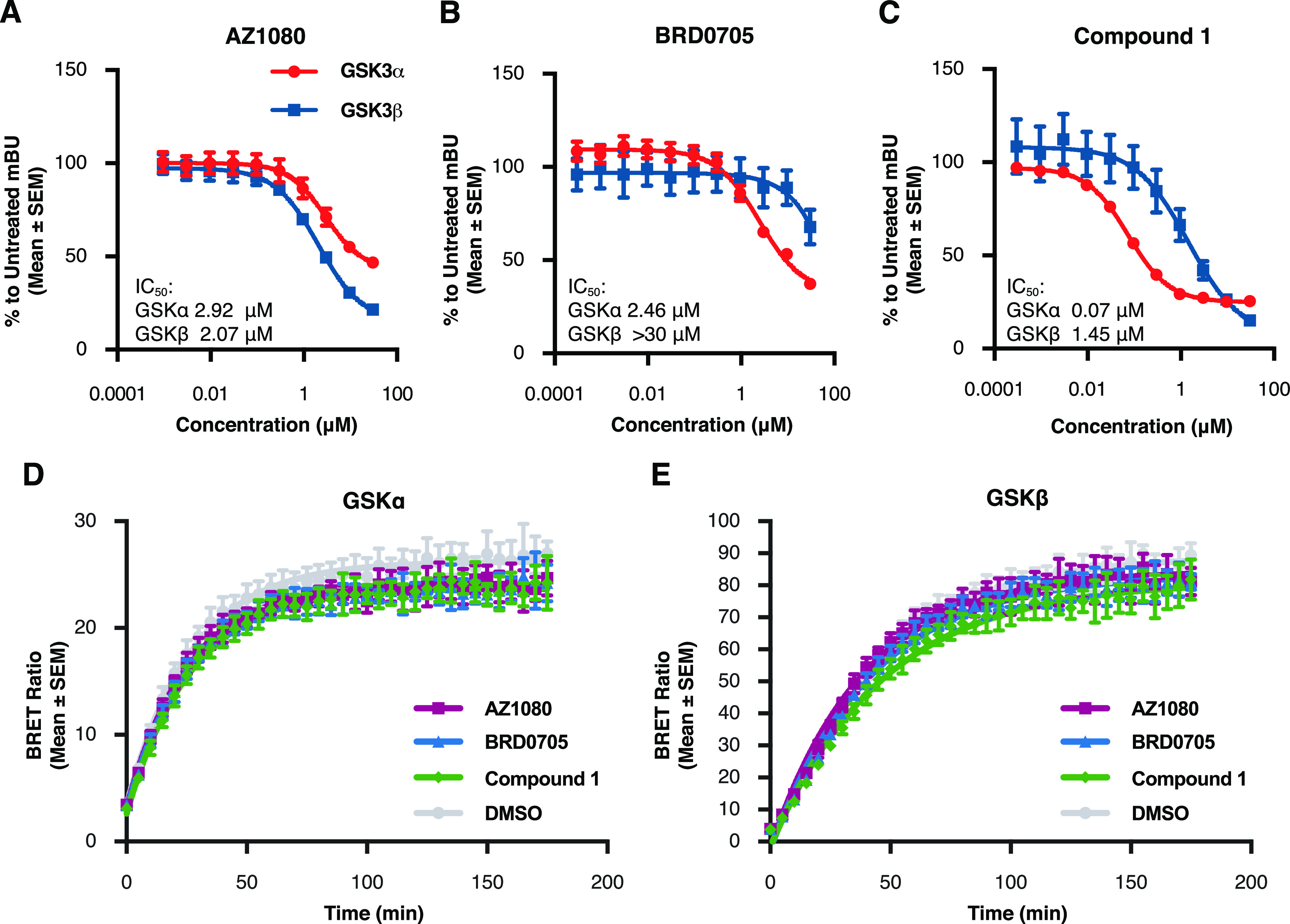
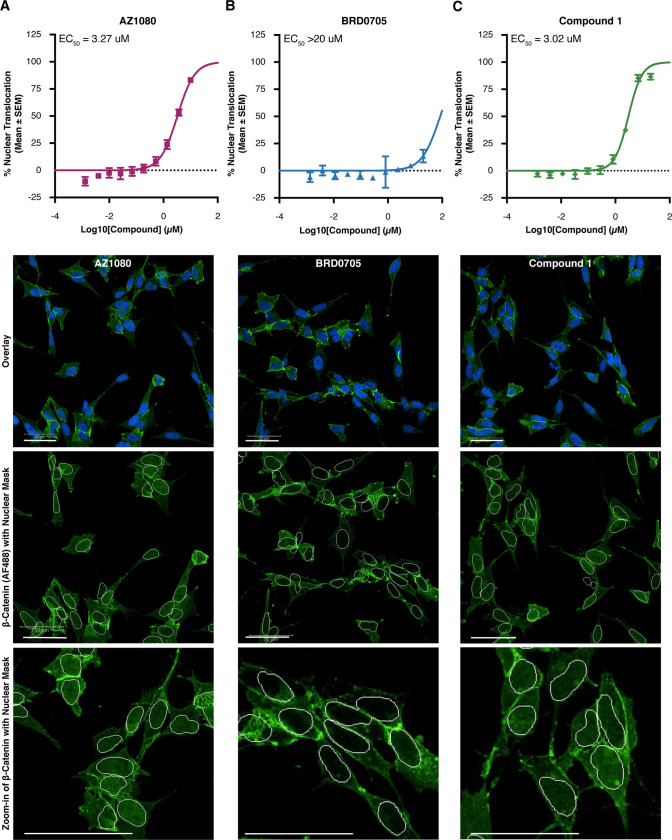
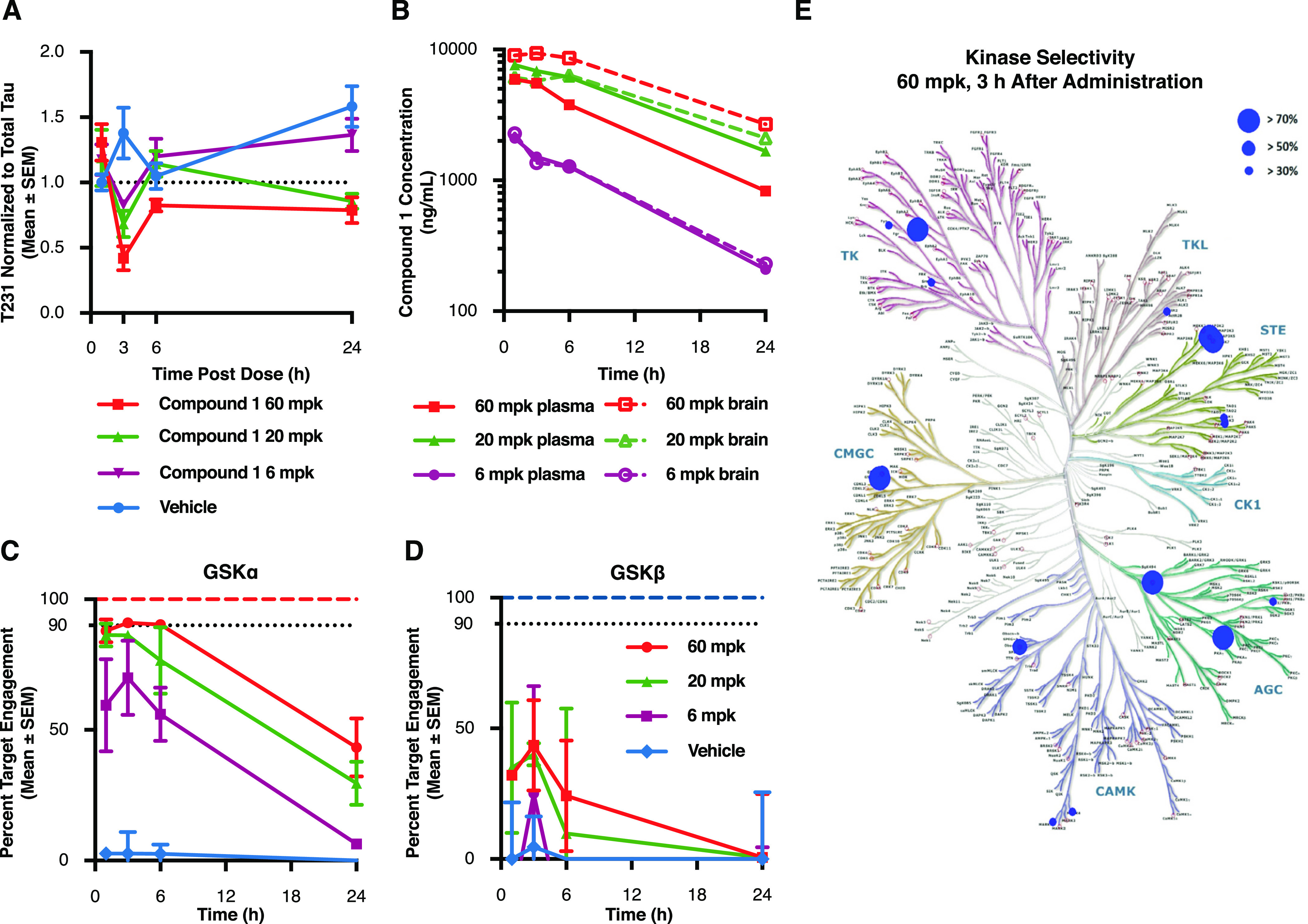
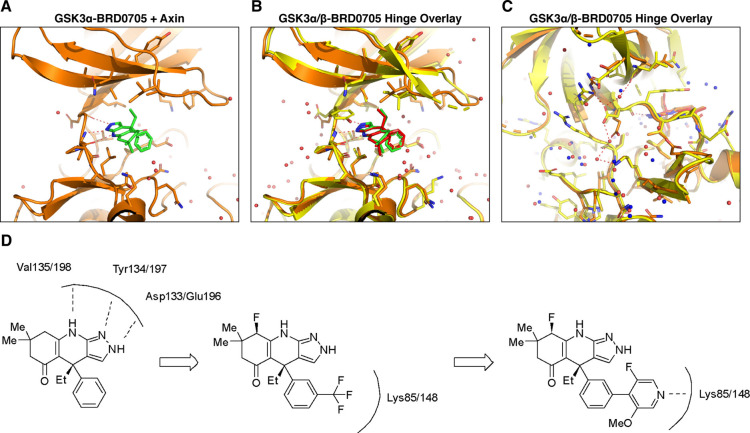
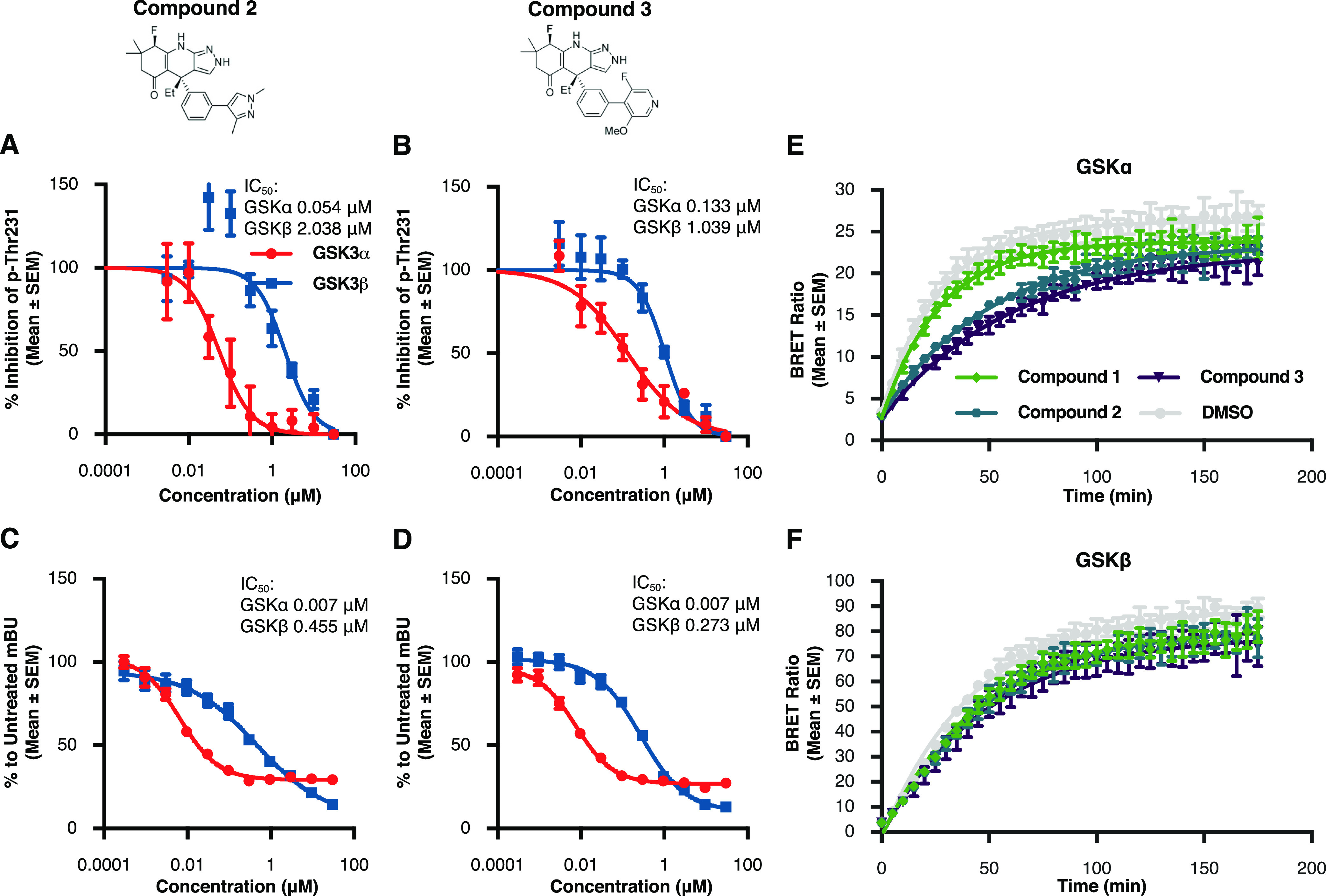
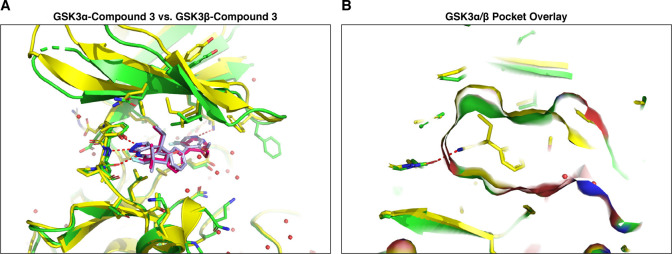
Glycogen synthase kinase 3 (GSK3) remains a therapeutic target of interest for diverse clinical indications. However, one hurdle in the development of small molecule GSK3 inhibitors has been safety concerns related to pan-inhibition of both GSK3 paralogs, leading to activation of the Wnt/β-catenin pathway and potential for aberrant cell proliferation. Development of GSK3α or GSK3β paralog-selective inhibitors that could offer an improved safety profile has been reported but further advancement has been hampered by the lack of structural information for GSK3α. Here we report for the first time the crystal structure for GSK3α, both in apo form and bound to a paralog-selective inhibitor. Taking advantage of this new structural information, we describe the design and in vitro testing of novel compounds with up to ∼37-fold selectivity for GSK3α over GSK3β with favorable drug-like properties. Furthermore, using chemoproteomics, we confirm that acute inhibition of GSK3α can lower tau phosphorylation at disease-relevant sites in vivo, with a high degree of selectivity over GSK3β and other kinases. Altogether, our studies advance prior efforts to develop GSK3 inhibitors by describing GSK3α structure and novel GSK3α inhibitors with improved selectivity, potency, and activity in disease-relevant systems.
Keywords: Alzheimer’s; GSK3α; GSK3β; kinase inhibitor; tau; β-catenin.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
Similar articles
-
Exploiting an Asp-Glu "switch" in glycogen synthase kinase 3 to design paralog-selective inhibitors for use in acute myeloid leukemia.Sci Transl Med. 2018 Mar 7;10(431):eaam8460. doi: 10.1126/scitranslmed.aam8460. Sci Transl Med. 2018. PMID: 29515000 Free PMC article.
-
GSK3β isoform-selective regulation of depression, memory and hippocampal cell proliferation.Genes Brain Behav. 2016 Mar;15(3):348-55. doi: 10.1111/gbb.12283. Epub 2016 Feb 12. Genes Brain Behav. 2016. PMID: 26749572 Free PMC article.
-
GSK3α: An Important Paralog in Neurodegenerative Disorders and Cancer.Biomolecules. 2020 Dec 16;10(12):1683. doi: 10.3390/biom10121683. Biomolecules. 2020. PMID: 33339170 Free PMC article. Review.
-
Glycogen synthase kinase 3 alpha and 3 beta have distinct functions during cardiogenesis of zebrafish embryo.BMC Dev Biol. 2007 Aug 3;7:93. doi: 10.1186/1471-213X-7-93. BMC Dev Biol. 2007. PMID: 17683539 Free PMC article.
-
Glycogen Synthase Kinase 3 (GSK3): Its Role and Inhibitors.Curr Top Med Chem. 2020;20(17):1522-1534. doi: 10.2174/1568026620666200516153136. Curr Top Med Chem. 2020. PMID: 32416693 Review.
Cited by
-
GSK3: A potential target and pending issues for treatment of Alzheimer's disease.CNS Neurosci Ther. 2024 Jul;30(7):e14818. doi: 10.1111/cns.14818. CNS Neurosci Ther. 2024. PMID: 38946682 Free PMC article. Review.
-
Recent advances in Alzheimer's disease: Mechanisms, clinical trials and new drug development strategies.Signal Transduct Target Ther. 2024 Aug 23;9(1):211. doi: 10.1038/s41392-024-01911-3. Signal Transduct Target Ther. 2024. PMID: 39174535 Free PMC article. Review.
-
GSK3A promotes human adenovirus replication and phosphorylates viral L4-22K protein.Life Sci Alliance. 2025 Jun 19;8(9):e202503320. doi: 10.26508/lsa.202503320. Print 2025 Sep. Life Sci Alliance. 2025. PMID: 40537285 Free PMC article.
References
-
- Bhat R.; Xue Y.; Berg S.; Hellberg S.; Ormö M.; Nilsson Y.; Radesäter A. C.; Jerning E.; Markgren P. O.; Borgegård T.; Nylöf M.; Giménez-Cassina A.; Hernández F.; Lucas J. J.; Díaz-Nido J.; Avila J. Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418. J. Biol. Chem. 2003, 278, 45937–45945. 10.1074/jbc.M306268200. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
