

Identifying key multifunctional components shared by critical cancer and normal liver pathways via SparseGMM
- PMID: 36814838
- PMCID: PMC9939431
- DOI: 10.1016/j.crmeth.2022.100392
Identifying key multifunctional components shared by critical cancer and normal liver pathways via SparseGMM
Abstract
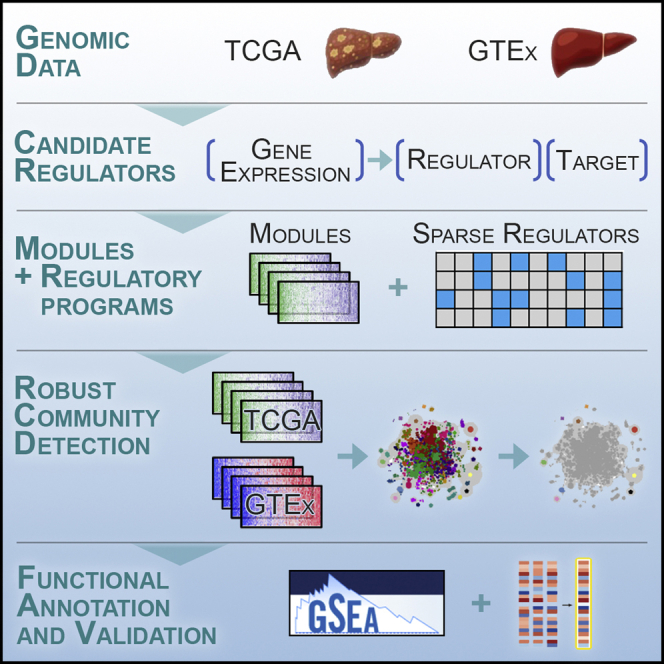
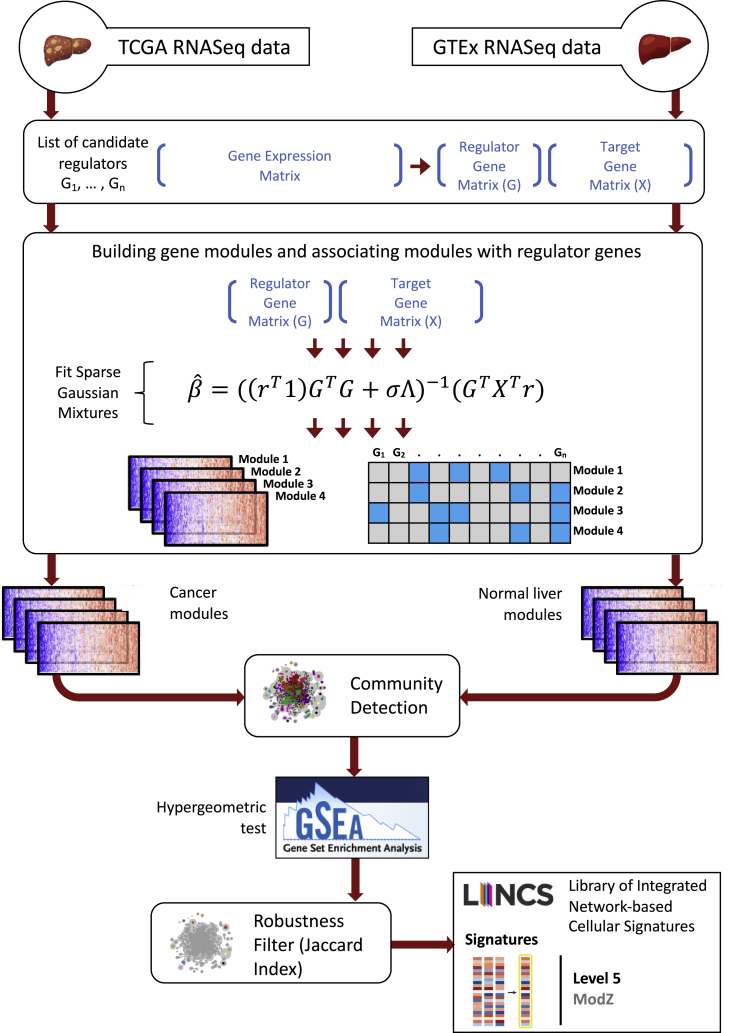
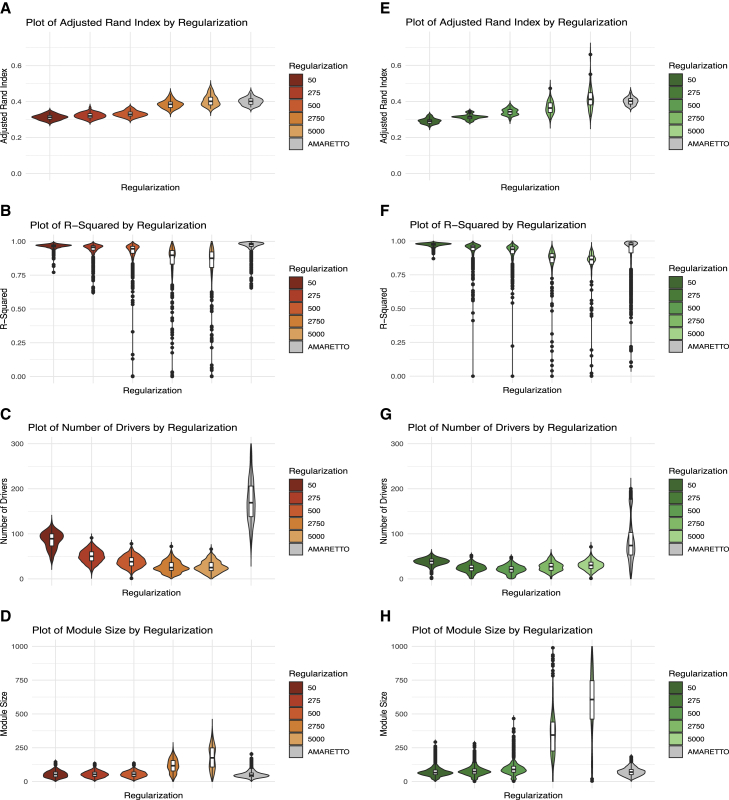
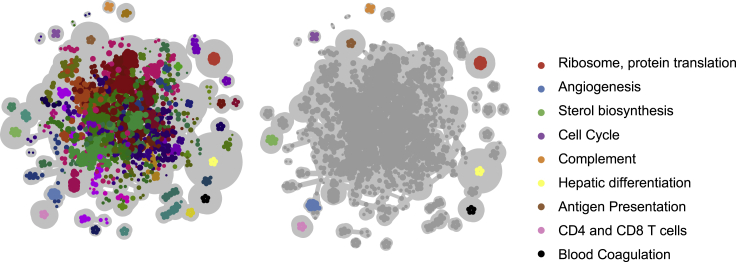
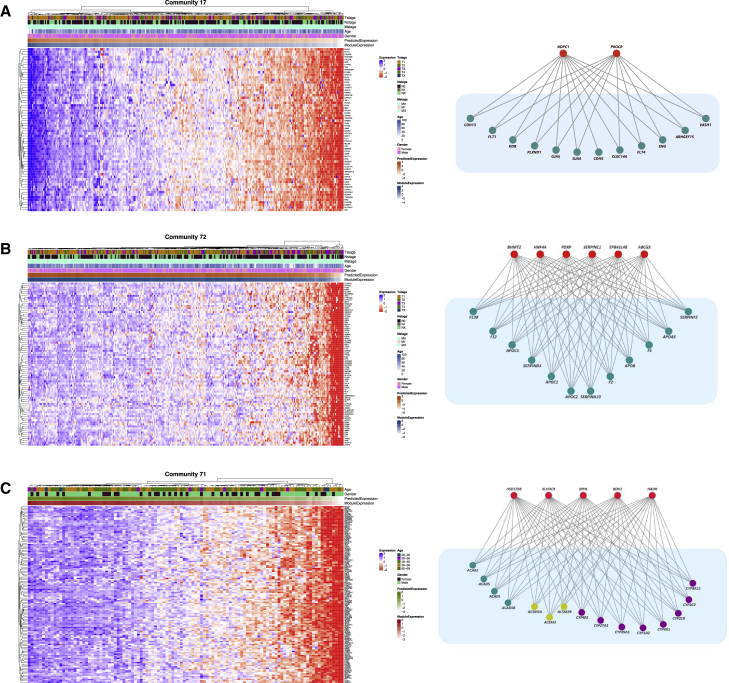
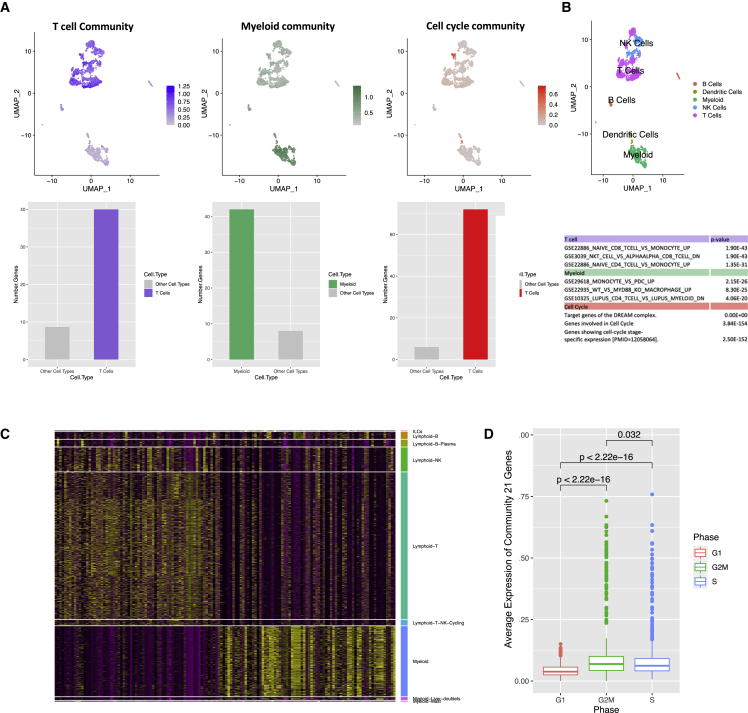
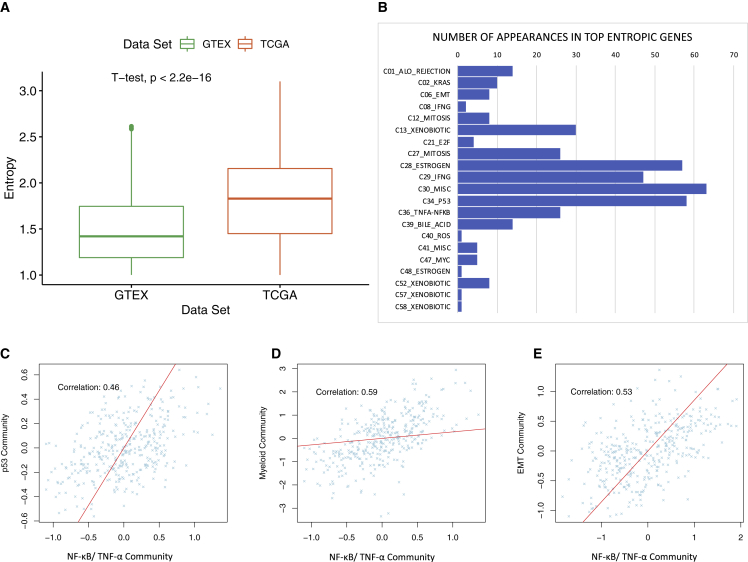
Despite the abundance of multimodal data, suitable statistical models that can improve our understanding of diseases with genetic underpinnings are challenging to develop. Here, we present SparseGMM, a statistical approach for gene regulatory network discovery. SparseGMM uses latent variable modeling with sparsity constraints to learn Gaussian mixtures from multiomic data. By combining coexpression patterns with a Bayesian framework, SparseGMM quantitatively measures confidence in regulators and uncertainty in target gene assignment by computing gene entropy. We apply SparseGMM to liver cancer and normal liver tissue data and evaluate discovered gene modules in an independent single-cell RNA sequencing (scRNA-seq) dataset. SparseGMM identifies PROCR as a regulator of angiogenesis and PDCD1LG2 and HNF4A as regulators of immune response and blood coagulation in cancer. Furthermore, we show that more genes have significantly higher entropy in cancer compared with normal liver. Among high-entropy genes are key multifunctional components shared by critical pathways, including p53 and estrogen signaling.
© 2022 The Authors.
Conflict of interest statement
The authors declare no competing interests
Figures
References
-
- GTEx Consortium. Laboratory, Data Analysis & Coordinating Center (LDACC)—Analysis Working Group. Statistical Methods groups-Analysis Working Group. Enhancing GTEx (eGTEx) groups. NIH Common Fund. NIH/NCI. NIH/NHGRI. NIH/NIMH. NIH/NIDA Genetic effects on gene expression across human tissues. Nature. 2017;550:204–213. et al. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
