

Mycobacterium tuberculosis infection triggers epigenetic changes that are enriched in a type I IFN signature
- PMID: 36820061
- PMCID: PMC9936219
- DOI: 10.1093/femsml/uqad006
Mycobacterium tuberculosis infection triggers epigenetic changes that are enriched in a type I IFN signature
Abstract
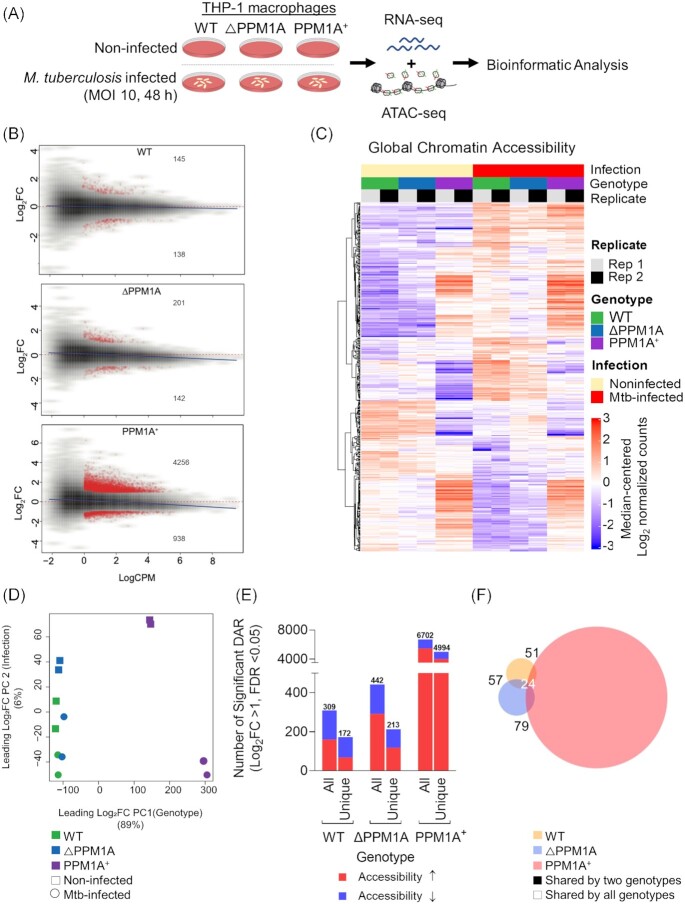
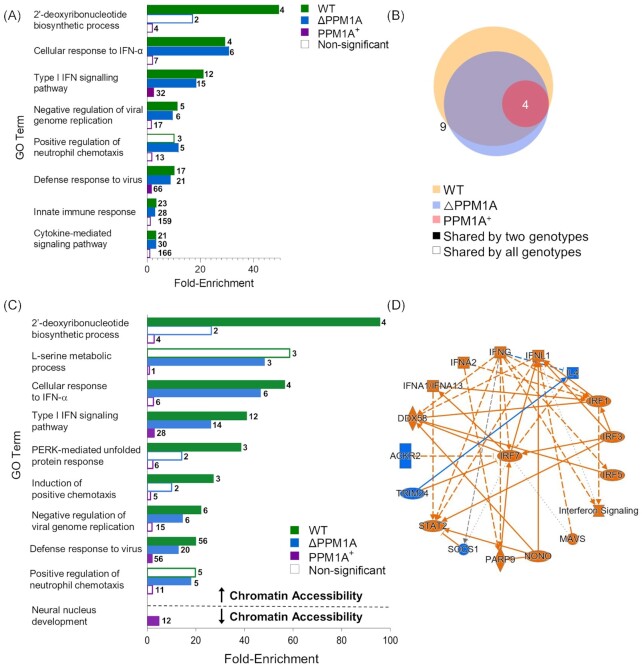
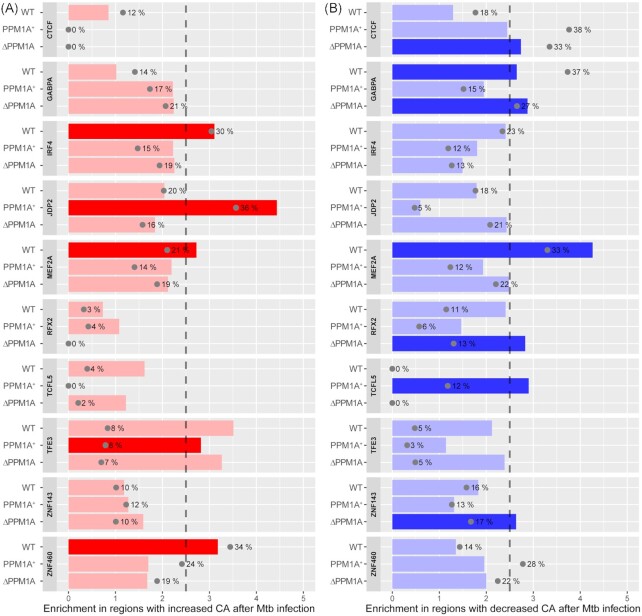
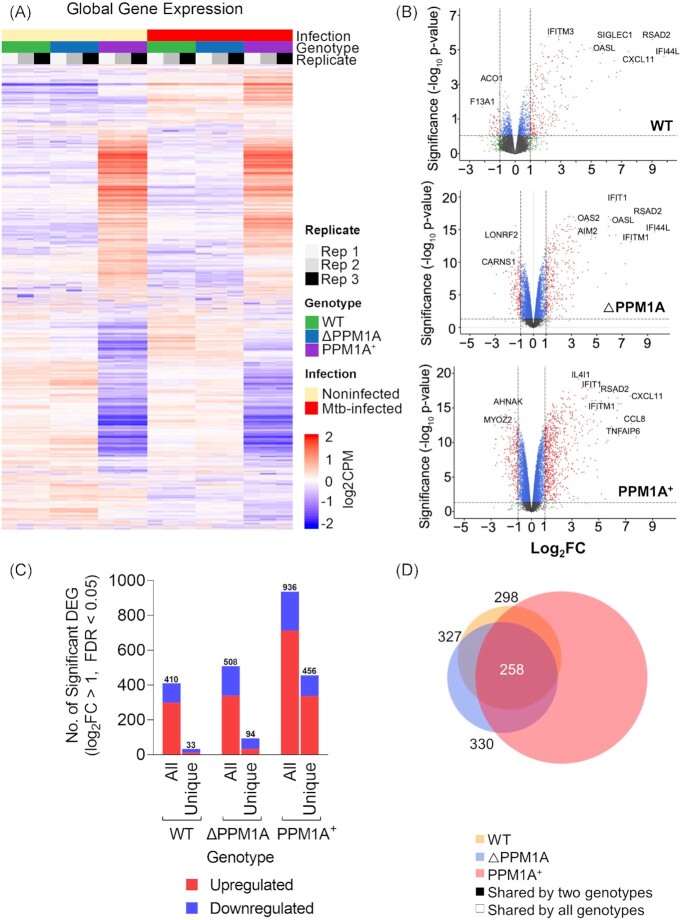
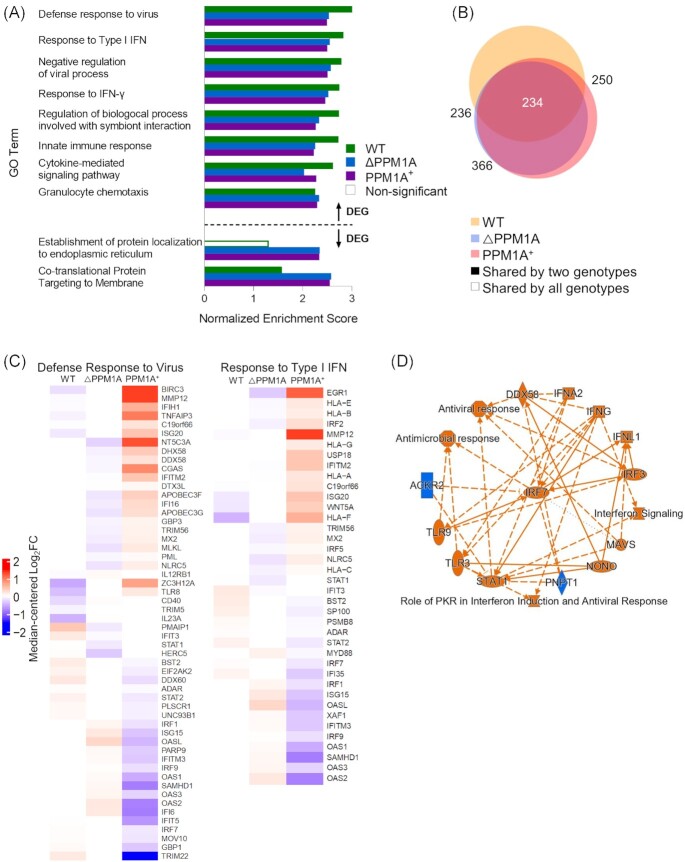
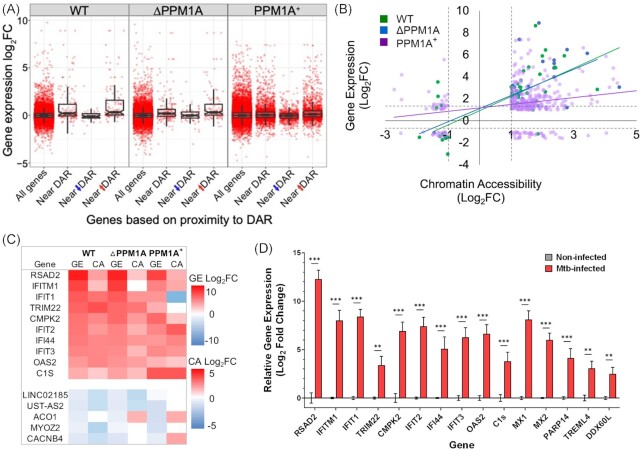
Tuberculosis, a deadly infectious lung disease caused by Mycobacterium tuberculosis (Mtb), remains the leading cause of bacterial disease-related deaths worldwide. Mtb reprograms and disables key antibacterial response pathways, many of which are regulated by epigenetic mechanisms that control the accessibility of chromatin to the transcriptional machinery. Recent reports suggest that host phosphatases, such as PPM1A, contribute to regulating chromatin accessibility during bacterial infections. However, changes in genome-wide chromatin accessibility during Mtb infection and whether PPM1A plays a role in this process remains unknown. Herein, we use combinatorial chromatin accessibility (ATAC-seq) and transcriptomic (RNA-seq) profiling of wild-type, PPM1A knockout and PPM1A overexpressing macrophages to demonstrate that Mtb infection induces global chromatin remodelling consistent with changes in gene expression. The strongest concordant changes to chromatin accessibility and gene expression triggered by Mtb infection were enriched for genes involved in type I interferon (IFN) signalling pathways. A panel of 15 genes with the strongest concordant changes in chromatin accessibility and gene expression were validated to be significantly upregulated in Mtb-infected human monocyte-derived macrophages. PPM1A expression affects chromatin accessibility profiles during Mtb infection that are reflected in the total number, chromosome location, and directionality of change. Transcription factor binding motif analysis revealed enrichment for transcription factors involved in the type I IFN pathway during Mtb infection, including members of the IRF, MEF2, and AP-1 families. Our study shows that altered type I IFN responses in Mtb-infected macrophages occur due to genome-wide changes in chromatin accessibility, and that PPM1A could influence a subset of these signatures.
Keywords: ATAC-seq; Mycobacterium tuberculosis; RNA-seq; chromatin accessibility; epigenome; host-directed therapy; transcriptome; tuberculosis; type I IFN.
© The Author(s) 2023. Published by Oxford University Press on behalf of FEMS.
Conflict of interest statement
None declared.
Figures
References
-
- Berton S, Chen L, Liang YC, et al. . A selective PPM1A inhibitor activates autophagy to restrict the survival of Mycobacterium tuberculosis. Cell Chem Biol. 2022;29:1126–39.e12. - PubMed
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials