

MacroH2A histone variants modulate enhancer activity to repress oncogenic programs and cellular reprogramming
- PMID: 36823213
- PMCID: PMC9950461
- DOI: 10.1038/s42003-023-04571-1
MacroH2A histone variants modulate enhancer activity to repress oncogenic programs and cellular reprogramming
Abstract
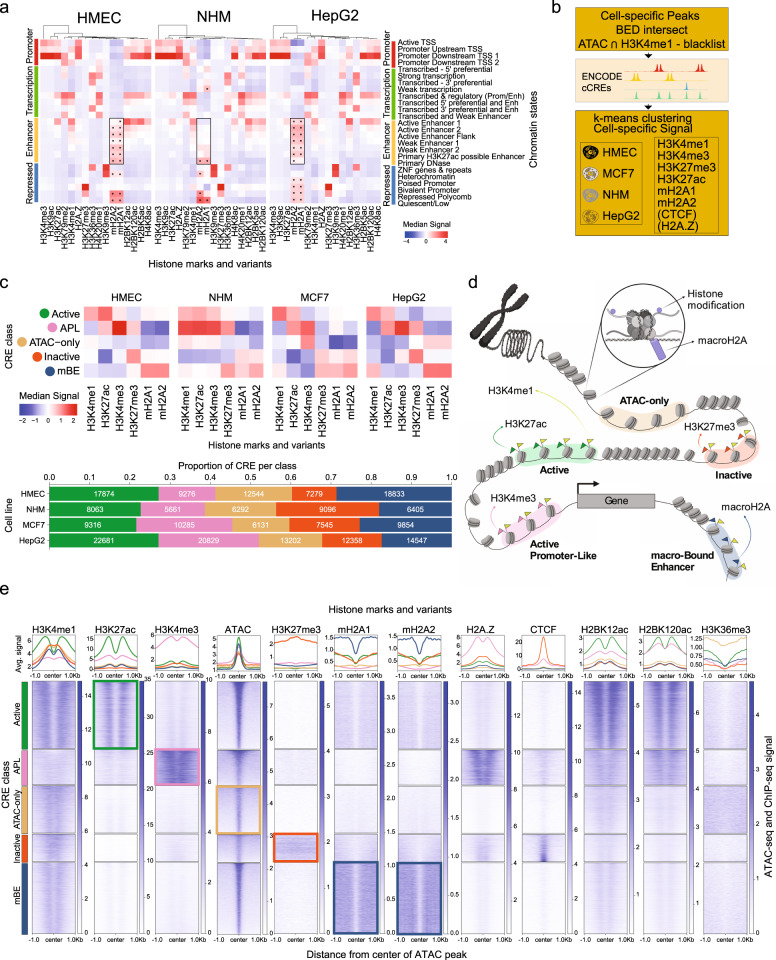
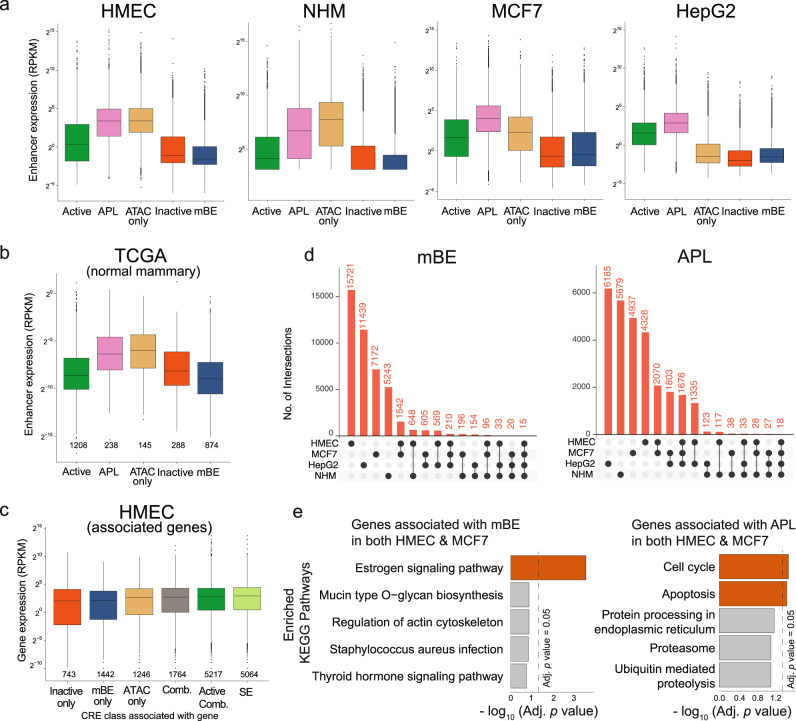
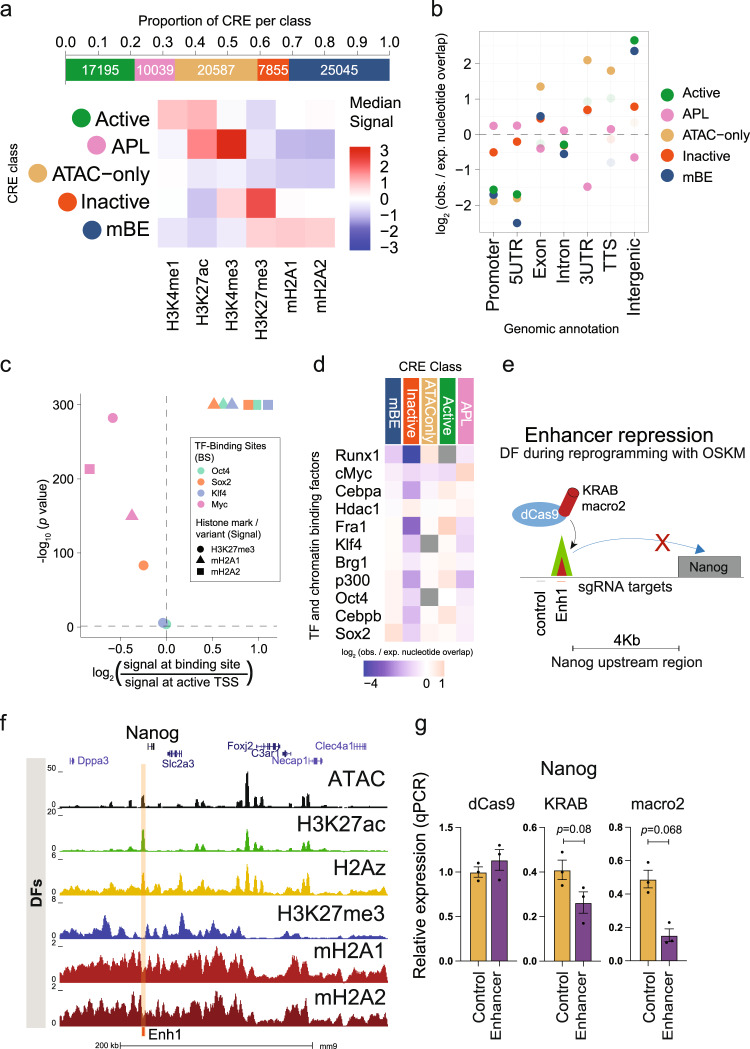
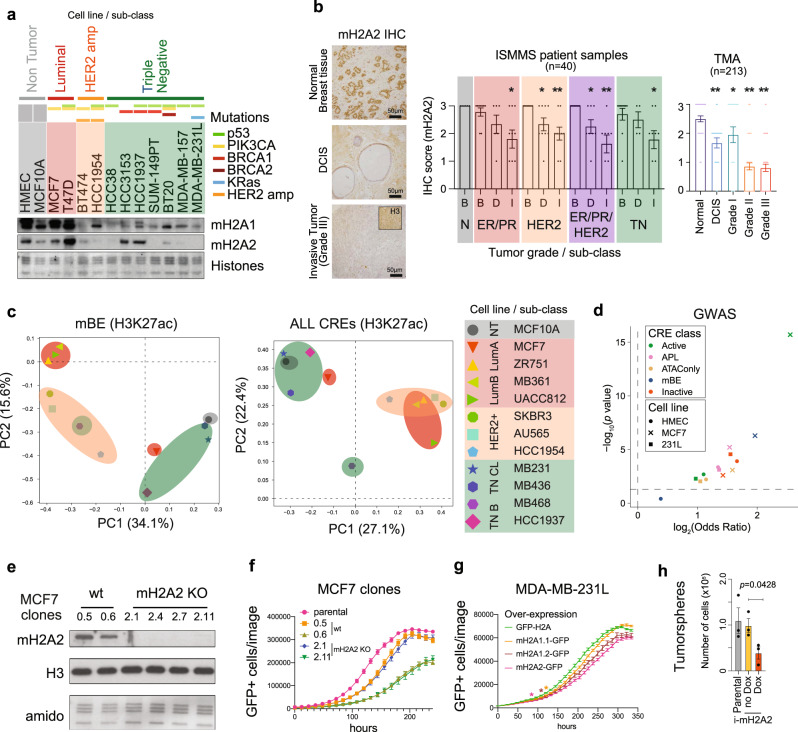
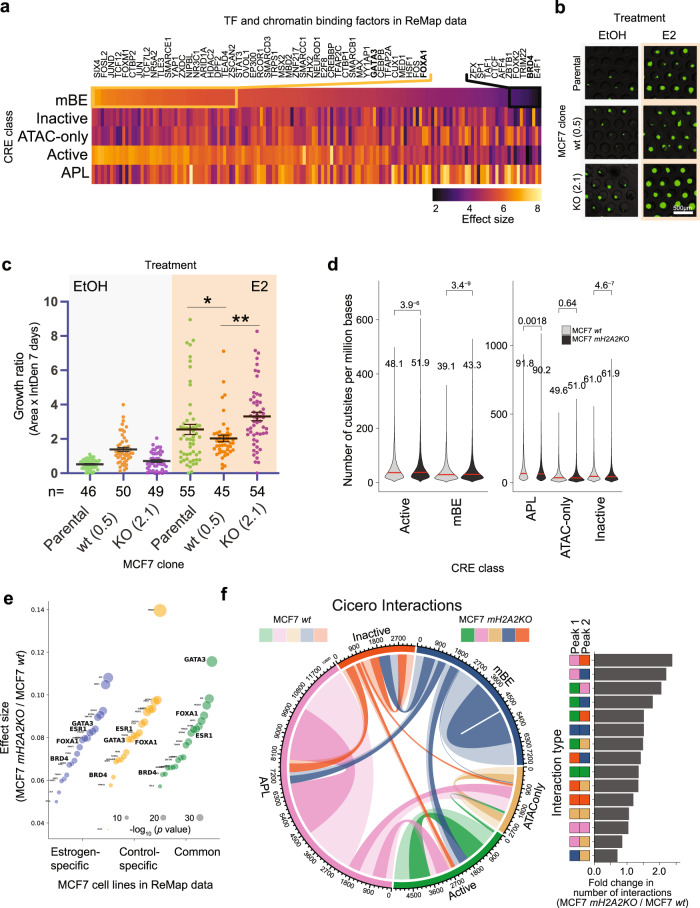
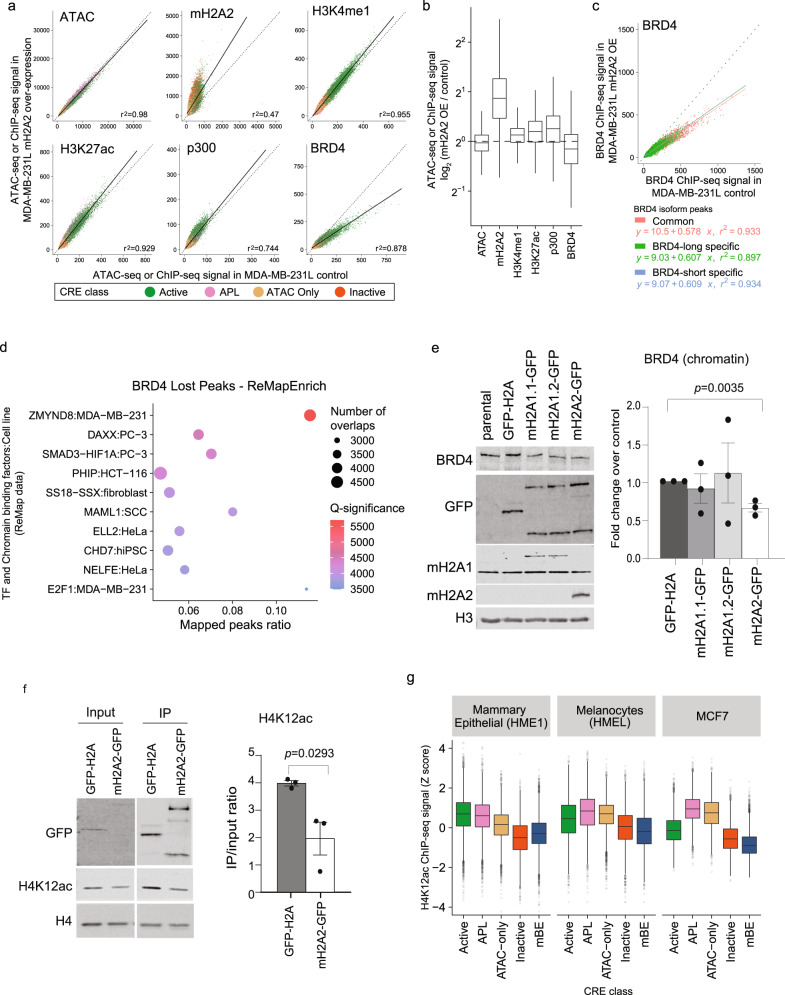
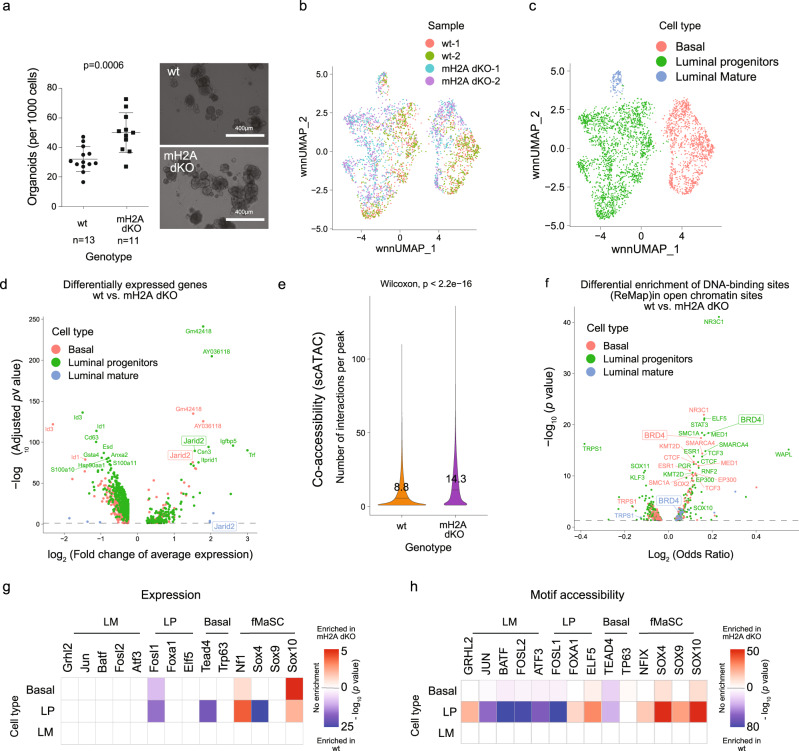
Considerable efforts have been made to characterize active enhancer elements, which can be annotated by accessible chromatin and H3 lysine 27 acetylation (H3K27ac). However, apart from poised enhancers that are observed in early stages of development and putative silencers, the functional significance of cis-regulatory elements lacking H3K27ac is poorly understood. Here we show that macroH2A histone variants mark a subset of enhancers in normal and cancer cells, which we coined 'macro-Bound Enhancers', that modulate enhancer activity. We find macroH2A variants localized at enhancer elements that are devoid of H3K27ac in a cell type-specific manner, indicating a role for macroH2A at inactive enhancers to maintain cell identity. In following, reactivation of macro-bound enhancers is associated with oncogenic programs in breast cancer and their repressive role is correlated with the activity of macroH2A2 as a negative regulator of BRD4 chromatin occupancy. Finally, through single cell epigenomic profiling of normal mammary stem cells derived from mice, we show that macroH2A deficiency facilitates increased activity of transcription factors associated with stem cell activity.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
A unique chromatin signature uncovers early developmental enhancers in humans.Nature. 2011 Feb 10;470(7333):279-83. doi: 10.1038/nature09692. Epub 2010 Dec 15. Nature. 2011. PMID: 21160473 Free PMC article.
-
Disruption of BRD4 at H3K27Ac-enriched enhancer region correlates with decreased c-Myc expression in Merkel cell carcinoma.Epigenetics. 2015;10(6):460-6. doi: 10.1080/15592294.2015.1034416. Epub 2015 May 5. Epigenetics. 2015. PMID: 25941994 Free PMC article.
-
Histone H3K27ac separates active from poised enhancers and predicts developmental state.Proc Natl Acad Sci U S A. 2010 Dec 14;107(50):21931-6. doi: 10.1073/pnas.1016071107. Epub 2010 Nov 24. Proc Natl Acad Sci U S A. 2010. PMID: 21106759 Free PMC article.
-
Epigenetic plasticity of enhancers in cancer.Transcription. 2020 Feb;11(1):26-36. doi: 10.1080/21541264.2020.1713682. Epub 2020 Jan 16. Transcription. 2020. PMID: 31944157 Free PMC article. Review.
-
Oncogenic super-enhancers in cancer: mechanisms and therapeutic targets.Cancer Metastasis Rev. 2023 Jun;42(2):471-480. doi: 10.1007/s10555-023-10103-4. Epub 2023 Apr 14. Cancer Metastasis Rev. 2023. PMID: 37059907 Free PMC article. Review.
Cited by
-
Combinatorial targeting of a specific EMT/MET network by macroH2A variants safeguards mesenchymal identity.PLoS One. 2023 Jul 11;18(7):e0288005. doi: 10.1371/journal.pone.0288005. eCollection 2023. PLoS One. 2023. PMID: 37432970 Free PMC article.
-
Melanoma and its fibroblastic allies: the emerging importance of CAFs in immune suppression, ECM modulation, and therapy resistance.Naunyn Schmiedebergs Arch Pharmacol. 2025 Aug 28. doi: 10.1007/s00210-025-04565-2. Online ahead of print. Naunyn Schmiedebergs Arch Pharmacol. 2025. PMID: 40874993 Review.
-
Roles of Histone H2A Variants in Cancer Development, Prognosis, and Treatment.Int J Mol Sci. 2024 Mar 9;25(6):3144. doi: 10.3390/ijms25063144. Int J Mol Sci. 2024. PMID: 38542118 Free PMC article. Review.
-
Multifunctional histone variants in genome function.Nat Rev Genet. 2025 Feb;26(2):82-104. doi: 10.1038/s41576-024-00759-1. Epub 2024 Aug 13. Nat Rev Genet. 2025. PMID: 39138293 Review.
-
Characterization and Optimization of Multiomic Single-Cell Epigenomic Profiling.Genes (Basel). 2023 Jun 10;14(6):1245. doi: 10.3390/genes14061245. Genes (Basel). 2023. PMID: 37372428 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
