

Retrospective analysis of enhancer activity and transcriptome history
- PMID: 36823354
- PMCID: PMC10635829
- DOI: 10.1038/s41587-023-01683-1
Retrospective analysis of enhancer activity and transcriptome history
Abstract
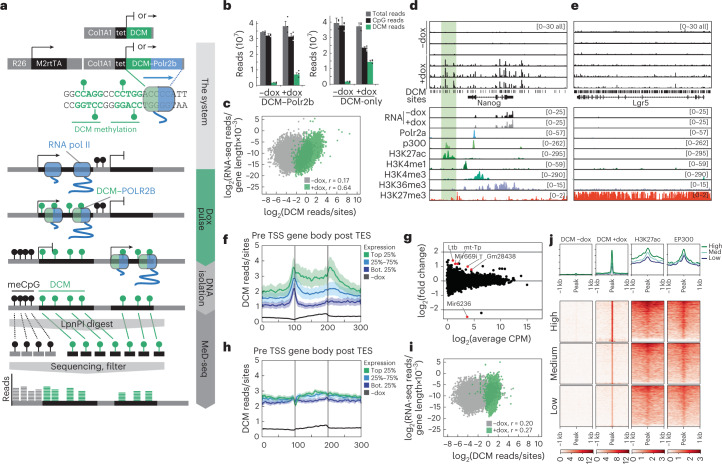
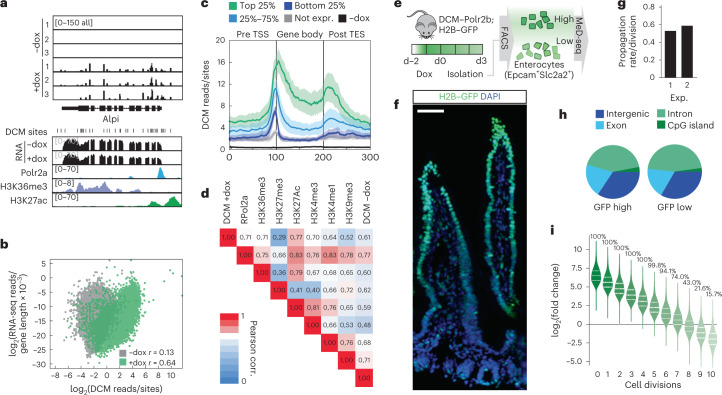
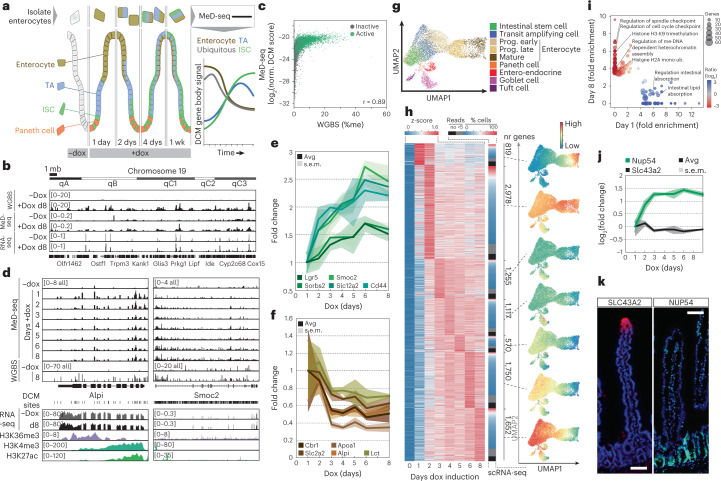
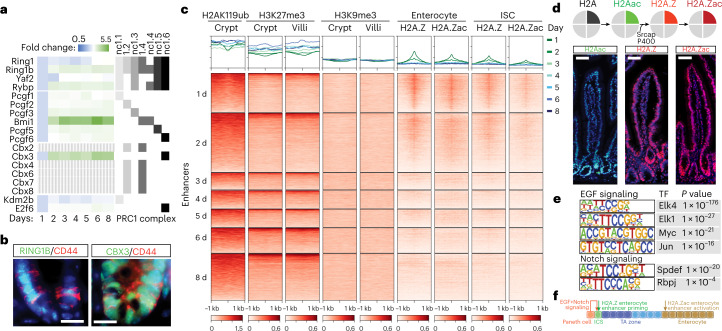
Cell state changes in development and disease are controlled by gene regulatory networks, the dynamics of which are difficult to track in real time. In this study, we used an inducible DCM-RNA polymerase subunit b fusion protein which labels active genes and enhancers with a bacterial methylation mark that does not affect gene transcription and is propagated in S-phase. This DCM-RNA polymerase fusion protein enables transcribed genes and active enhancers to be tagged and then examined at later stages of development or differentiation. We apply this DCM-time machine (DCM-TM) technology to study intestinal homeostasis, revealing rapid and coordinated activation of enhancers and nearby genes during enterocyte differentiation. We provide new insights in absorptive-secretory lineage decision-making in intestinal stem cell (ISC) differentiation and show that ISCs retain a unique chromatin landscape required to maintain ISC identity and delineate future expression of differentiation-associated genes. DCM-TM has wide applicability in tracking cell states, providing new insights in the regulatory networks underlying cell state changes.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no conflicts of interest except for R.B., J.B., W.F.J.v.I. and J.G. who report being shareholders in Methylomics B.V., a commercial company that applies MeD-seq to develop methylation markers for cancer staging.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
