

This is a preprint.
Single-strand mismatch and damage patterns revealed by single-molecule DNA sequencing
- PMID: 36824744
- PMCID: PMC9949150
- DOI: 10.1101/2023.02.19.526140
Single-strand mismatch and damage patterns revealed by single-molecule DNA sequencing
Update in
-
DNA mismatch and damage patterns revealed by single-molecule sequencing.Nature. 2024 Jun;630(8017):752-761. doi: 10.1038/s41586-024-07532-8. Epub 2024 Jun 12. Nature. 2024. PMID: 38867045 Free PMC article.
Abstract
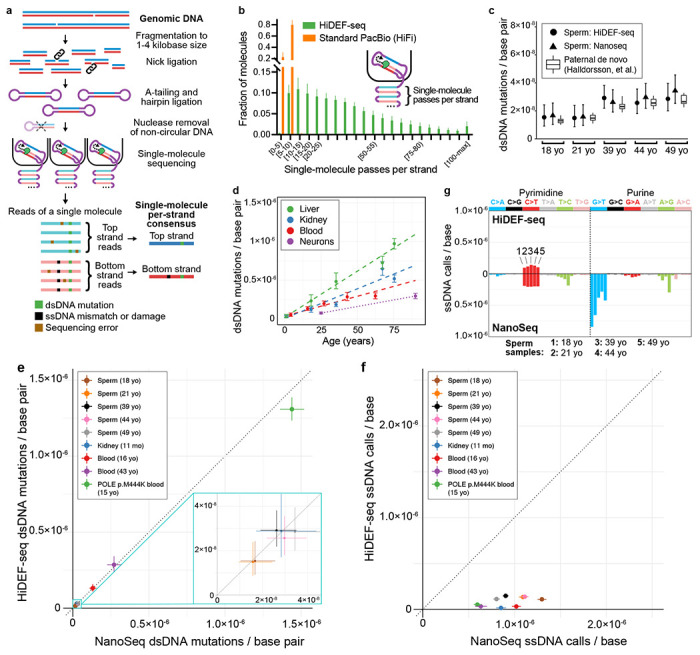
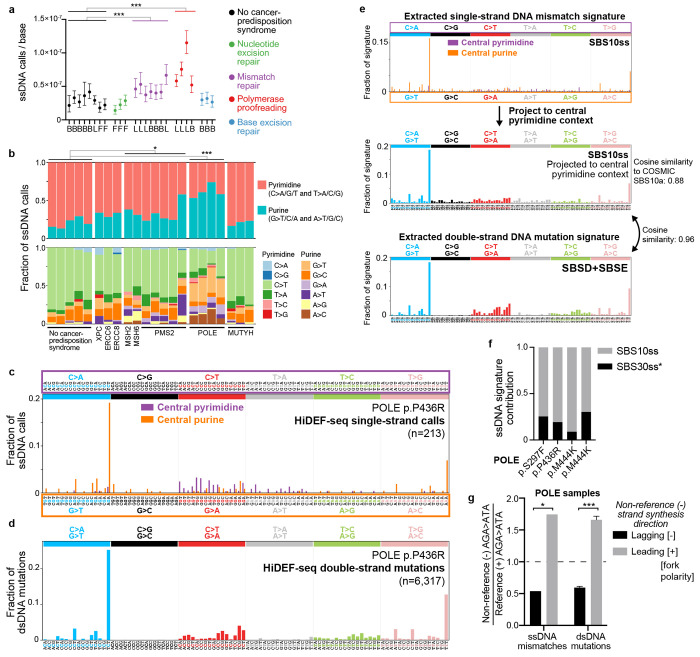
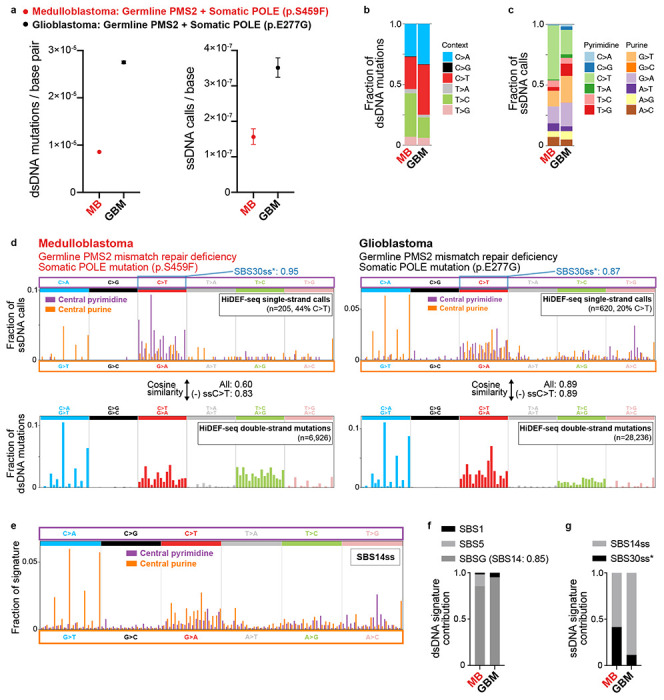
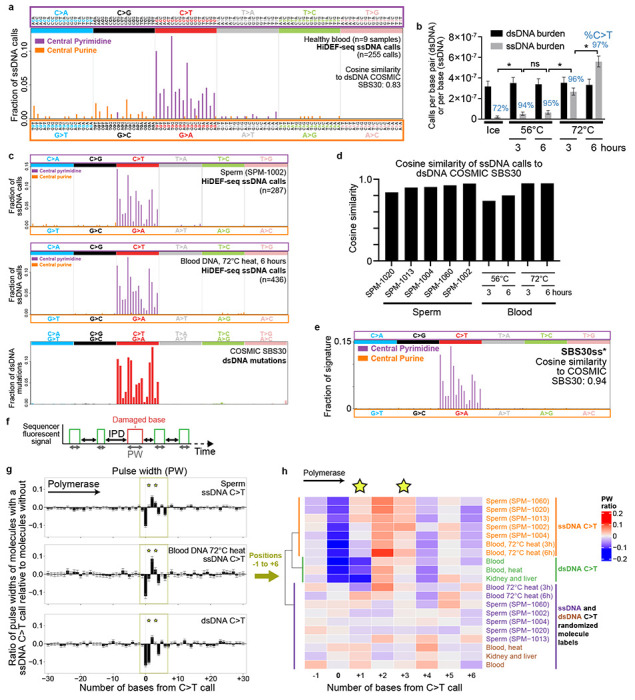
Mutations accumulate in the genome of every cell of the body throughout life, causing cancer and other genetic diseases1-4. Almost all of these mosaic mutations begin as nucleotide mismatches or damage in only one of the two strands of the DNA prior to becoming double-strand mutations if unrepaired or misrepaired5. However, current DNA sequencing technologies cannot resolve these initial single-strand events. Here, we developed a single-molecule, long-read sequencing method that achieves single-molecule fidelity for single-base substitutions when present in either one or both strands of the DNA. It also detects single-strand cytosine deamination events, a common type of DNA damage. We profiled 110 samples from diverse tissues, including from individuals with cancer-predisposition syndromes, and define the first single-strand mismatch and damage signatures. We find correspondences between these single-strand signatures and known double-strand mutational signatures, which resolves the identity of the initiating lesions. Tumors deficient in both mismatch repair and replicative polymerase proofreading show distinct single-strand mismatch patterns compared to samples deficient in only polymerase proofreading. In the mitochondrial genome, our findings support a mutagenic mechanism occurring primarily during replication. Since the double-strand DNA mutations interrogated by prior studies are only the endpoint of the mutation process, our approach to detect the initiating single-strand events at single-molecule resolution will enable new studies of how mutations arise in a variety of contexts, especially in cancer and aging.
Conflict of interest statement
Competing Interests A provisional patent application on HiDEF-seq has been filed (NYU Grossman School of Medicine). G.D.E. owns stock in DNA sequencing companies (Illumina, Oxford Nanopore Technologies, and Pacific Biosciences).
Figures


References
-
- Martincorena I. & Campbell P. J. Somatic mutation in cancer and normal cells. Science 349, 1483 (2015). - PubMed
-
- Mustjoki S. & Young N. S. Somatic Mutations in “Benign” Disease. New England Journal of Medicine 384, 2039–2052 (2021). - PubMed
-
- Moore L. et al. The mutational landscape of human somatic and germline cells. Nature 597, 381–386 (2021). - PubMed
-
- Seplyarskiy V. B. & Sunyaev S. The origin of human mutation in light of genomic data. Nature Reviews Genetics 22, 672–686 (2021). - PubMed
Methods References
-
- Agarwal A., Gupta S. & Sharma R. in Andrological Evaluation of Male Infertility: A Laboratory Guide (eds Agarwal Ashok, Gupta Sajal, & Sharma Rakesh) 101–107 (Springer International Publishing, 2016).
-
- Jenkins T. G., Liu L., Aston K. I. & Carrell D. T. Pre-screening method for somatic cell contamination in human sperm epigenetic studies. Systems Biology in Reproductive Medicine 64, 146–155 (2018). - PubMed
-
- Nurk S. et al. The complete sequence of a human genome. bioRxiv, 2021.2005.2026.445798 (2021).
-
- Heng L. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv (2013).
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials