

This is a preprint.
E2F1 Mediates SOX17 Deficiency-Induced Pulmonary Hypertension
- PMID: 36824855
- PMCID: PMC9949178
- DOI: 10.1101/2023.02.15.528740
E2F1 Mediates SOX17 Deficiency-Induced Pulmonary Hypertension
Update in
-
E2F1 Mediates SOX17 Deficiency-Induced Pulmonary Hypertension.Hypertension. 2023 Nov;80(11):2357-2371. doi: 10.1161/HYPERTENSIONAHA.123.21241. Epub 2023 Sep 22. Hypertension. 2023. PMID: 37737027 Free PMC article.
Abstract
Rationale: Rare genetic variants and genetic variation at loci in an enhancer in SRY-Box Transcription Factor 17 (SOX17) are identified in patients with idiopathic pulmonary arterial hypertension (PAH) and PAH with congenital heart disease. However, the exact role of genetic variants or mutation in SOX17 in PAH pathogenesis has not been reported.
Objectives: To investigate the role of SOX17 deficiency in pulmonary hypertension (PH) development.
Methods: Human lung tissue and endothelial cells (ECs) from IPAH patients were used to determine the expression of SOX17. Tie2Cre-mediated and EC-specific deletion of Sox17 mice were assessed for PH development. Single-cell RNA sequencing analysis, human lung ECs, and smooth muscle cell culture were performed to determine the role and mechanisms of SOX17 deficiency. A pharmacological approach was used in Sox17 deficiency mice for therapeutic implication.
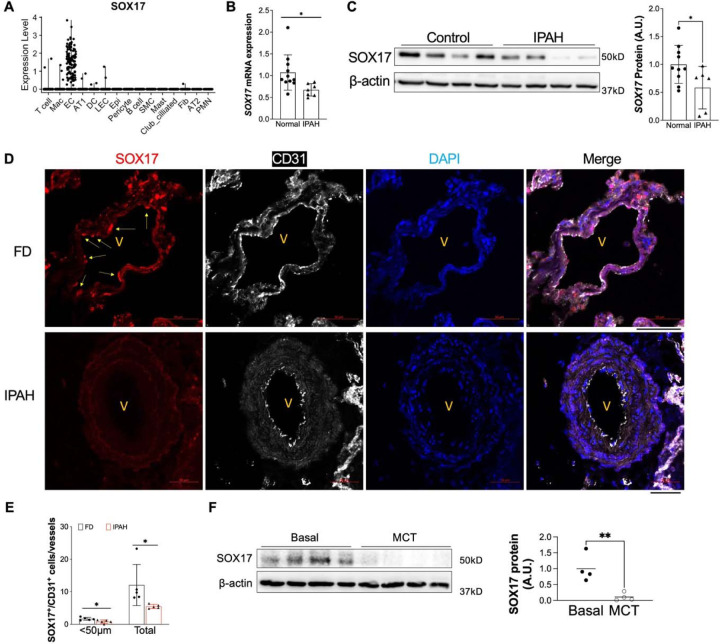
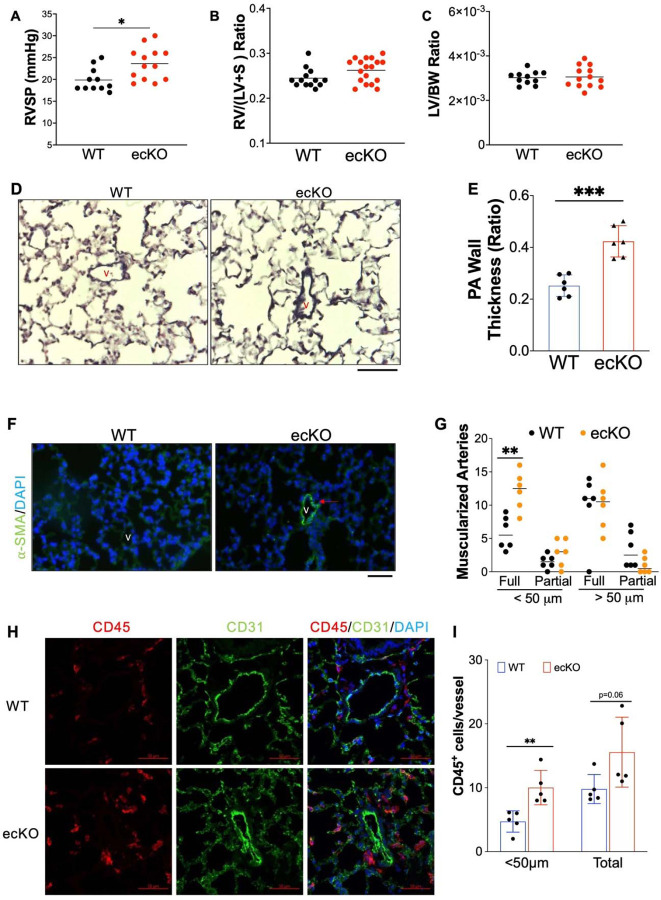
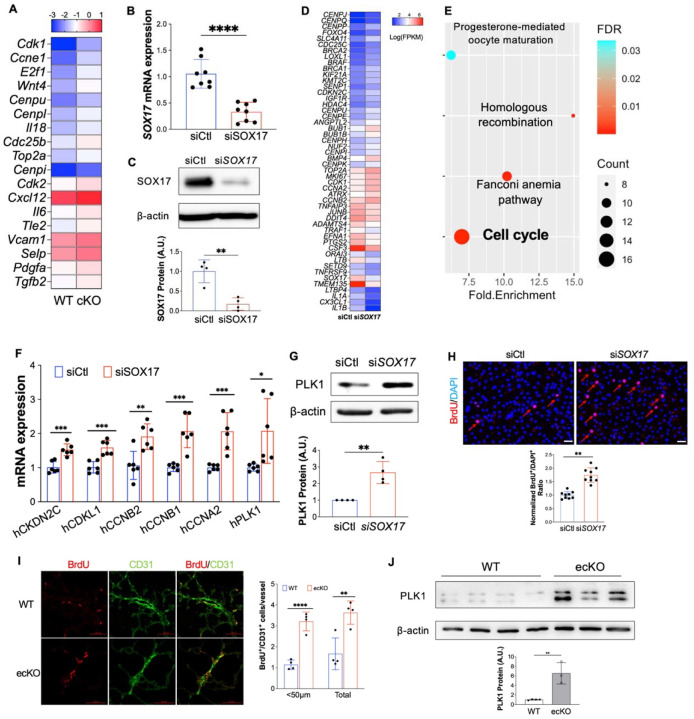
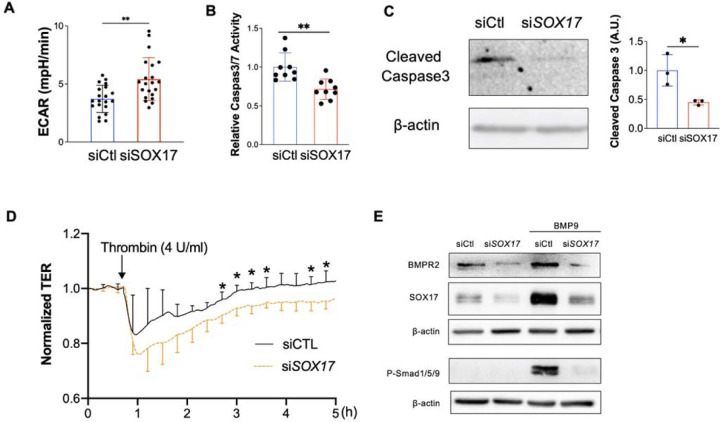
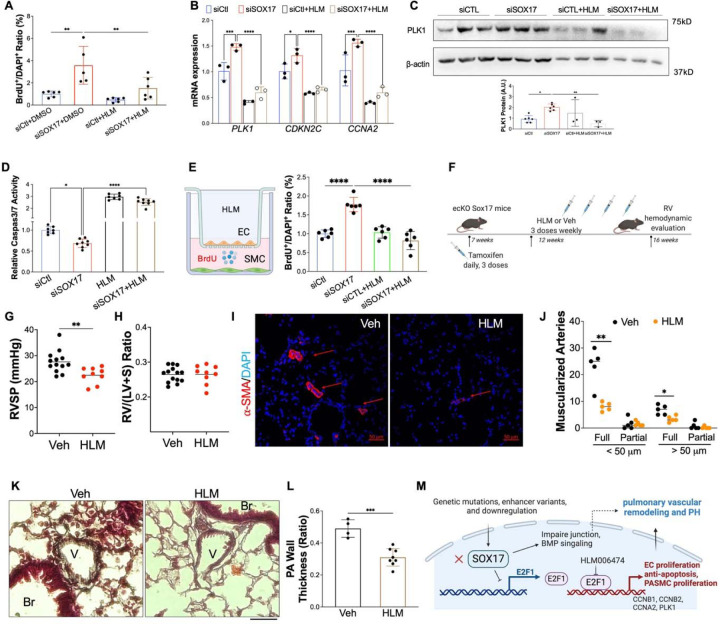
Measurement and main results: SOX17 expression was downregulated in the lungs and pulmonary ECs of IPAH patients. Mice with Tie2Cre mediated Sox17 knockdown and EC-specific Sox17 deletion developed spontaneously mild PH. Loss of endothelial Sox17 in EC exacerbated hypoxia-induced PH in mice. Loss of SOX17 in lung ECs induced endothelial dysfunctions including upregulation of cell cycle programming, proliferative and anti-apoptotic phenotypes, augmentation of paracrine effect on pulmonary arterial smooth muscle cells, impaired cellular junction, and BMP signaling. E2F Transcription Factor 1 (E2F1) signaling was shown to mediate the SOX17 deficiency-induced EC dysfunction and PH development.
Conclusions: Our study demonstrated that endothelial SOX17 deficiency induces PH through E2F1 and targeting E2F1 signaling represents a promising approach in PAH patients.
Figures




References
-
- Corris PA, Seeger W. Call it by the correct name-pulmonary hypertension not pulmonary arterial hypertension: Growing recognition of the global health impact for a well-recognized condition and the role of the Pulmonary Vascular Research Institute. Am J Physiol - Lung Cell Mol Physiol 2020;318:L992–L994. - PubMed
-
- Austin ED, Newman JH, Loyd JE, Phillips JA. Heritable and Idiopathic Forms of Pulmonary Arterial Hypertension. Emery Rimoin’s Princ Pract Med Genet Genomics, Seventh Ed. Elsevier; 2020. p. 439–462.doi: 10.1016/B978-0-12-812532-8.00016-1. - DOI
-
- Evans JDW, Girerd B, Montani D, Wang X-J, Galiè N, Austin ED, Elliott G, Asano K, Grünig E, Yan Y, Jing Z-C, Manes A, Palazzini M, Wheeler LA, Nakayama I, Satoh T, Eichstaedt C, Hinderhofer K, Wolf M, Rosenzweig EB, Chung WK, Soubrier F, Simonneau G, Sitbon O, Gräf S, Kaptoge S, Di Angelantonio E, Humbert M, Morrell NW. BMPR2 mutations and survival in pulmonary arterial hypertension: an individual participant data meta-analysis. Lancet Respir Med 2016;4:129–137. - PMC - PubMed
-
- Southgate L, Machado RD, Gräf S, Morrell NW. Molecular genetic framework underlying pulmonary arterial hypertension. Nat Rev Cardiol 2020;17:85–95. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources