

This is a preprint.
LPD-3 as a megaprotein brake for aging and insulin-mTOR signaling in C. elegans
- PMID: 36824874
- PMCID: PMC9949100
- DOI: 10.1101/2023.02.14.528431
LPD-3 as a megaprotein brake for aging and insulin-mTOR signaling in C. elegans
Update in
-
LPD-3 as a megaprotein brake for aging and insulin-mTOR signaling in C. elegans.Cell Rep. 2024 Mar 26;43(3):113899. doi: 10.1016/j.celrep.2024.113899. Epub 2024 Mar 5. Cell Rep. 2024. PMID: 38446666 Free PMC article.
Abstract
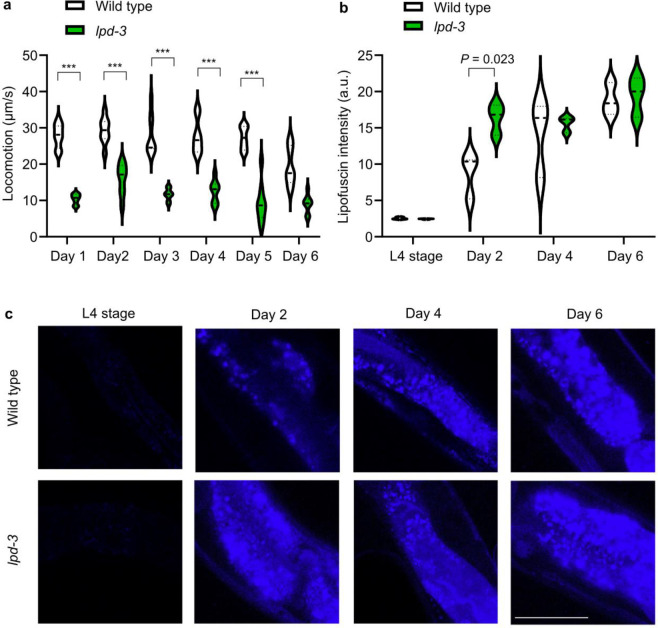
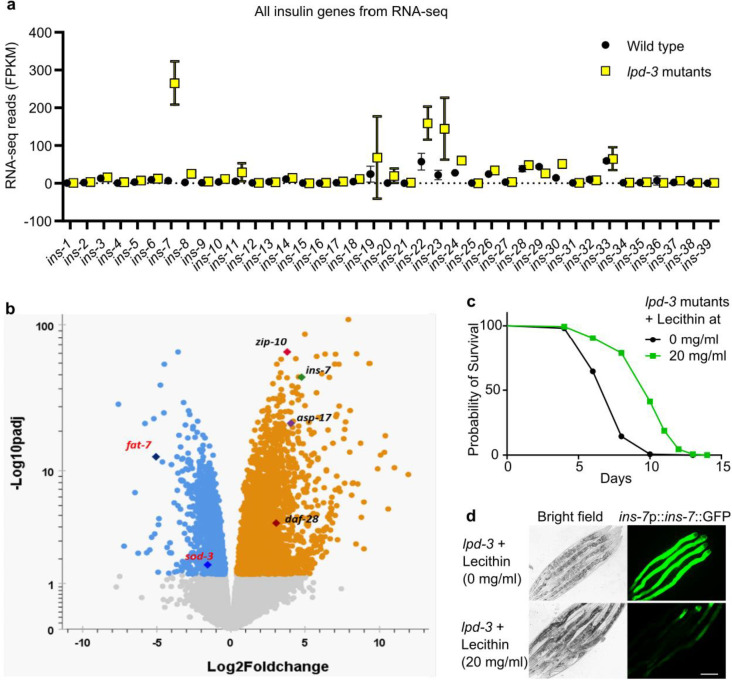
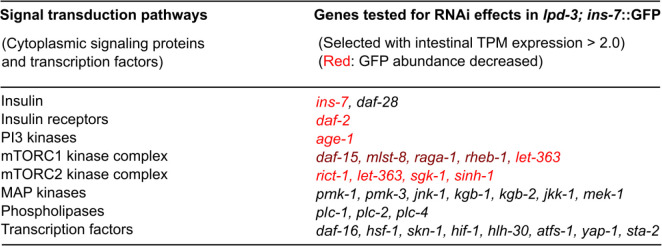
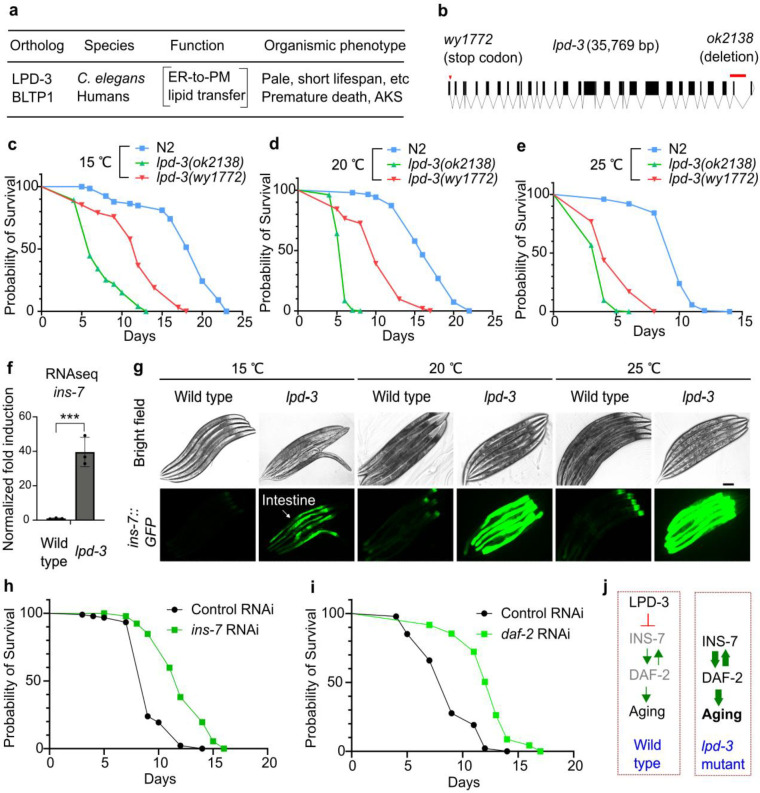
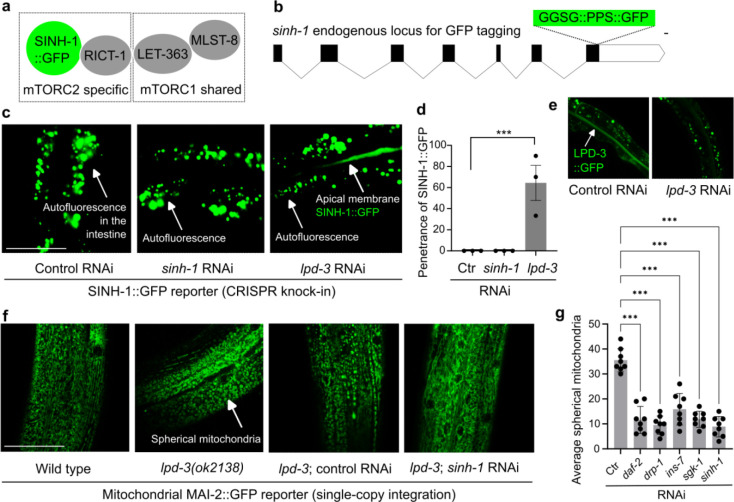
Insulin-mTOR signaling drives anabolic growth during organismal development, while its late-life dysregulation may detrimentally contribute to aging and limit lifespans. Age-related regulatory mechanisms and functional consequences of insulin-mTOR remain incompletely understood. Here we identify LPD-3 as a megaprotein that orchestrates the tempo of insulin-mTOR signaling during C. elegans aging. We find that an agonist insulin INS-7 is drastically over-produced in early life and shortens lifespan in lpd-3 mutants, a C. elegans model of human Alkuraya-Kučinskas syndrome. LPD-3 forms a bridge-like tunnel megaprotein to facilitate phospholipid trafficking to plasma membranes. Lipidomic profiling reveals increased abundance of hexaceramide species in lpd-3 mutants, accompanied by up-regulation of hexaceramide biosynthetic enzymes, including HYL-1 (Homolog of Yeast Longevity). Reducing HYL-1 activity decreases INS-7 levels and rescues the lifespan of lpd-3 mutants through insulin receptor/DAF-2 and mTOR/LET-363. LPD3 antagonizes SINH-1, a key mTORC2 component, and decreases expression with age in wild type animals. We propose that LPD-3 acts as a megaprotein brake for aging and its age-dependent decline restricts lifespan through the sphingolipid-hexaceramide and insulin-mTOR pathways.
Conflict of interest statement
Competing interests The authors declare no competing interests.
Figures



References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous