

This is a preprint.
Mitochondrial Pyruvate Carrier Inhibition Attenuates Hepatic Stellate Cell Activation and Liver Injury in a Mouse Model of Metabolic Dysfunction-associated Steatotic Liver Disease
- PMID: 36824926
- PMCID: PMC9949033
- DOI: 10.1101/2023.02.13.528384
Mitochondrial Pyruvate Carrier Inhibition Attenuates Hepatic Stellate Cell Activation and Liver Injury in a Mouse Model of Metabolic Dysfunction-associated Steatotic Liver Disease
Abstract
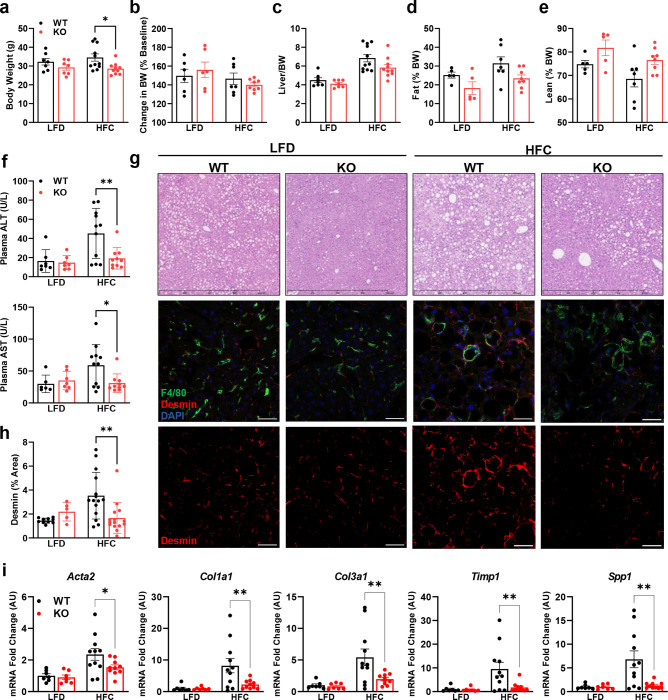
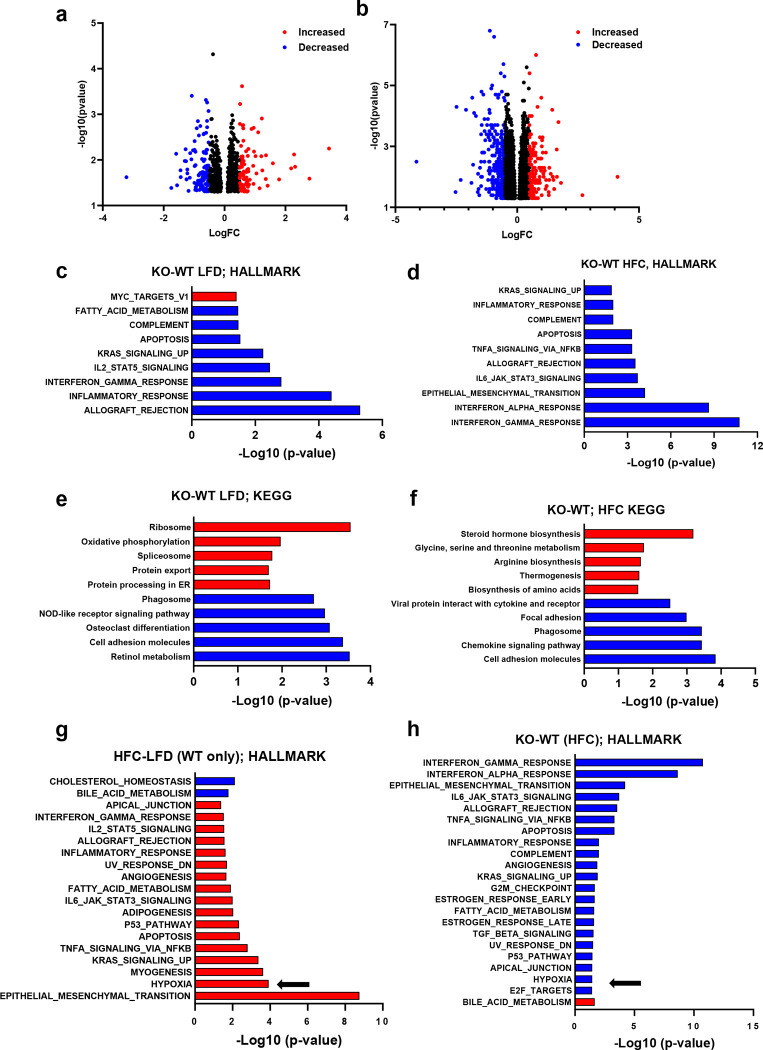
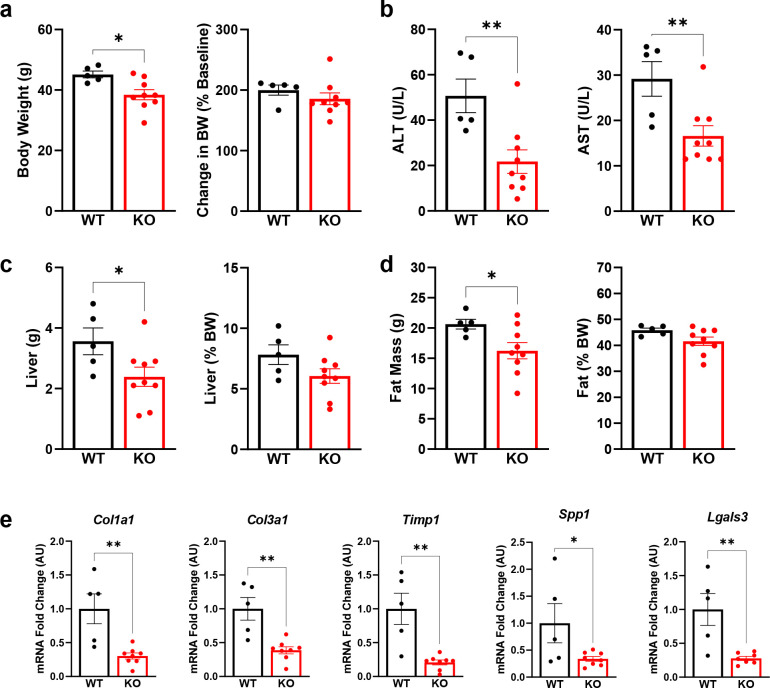
Hepatic stellate cells (HSC) are non-parenchymal liver cells that produce extracellular matrix comprising fibrotic lesions in chronic liver diseases. Prior work demonstrated that mitochondrial pyruvate carrier (MPC) inhibitors suppress HSC activation and fibrosis in a mouse model of metabolic dysfunction-associated steatohepatitis (MASH). In the present study, pharmacologic or genetic inhibition of the MPC in HSC decreased expression of markers of activation in vitro. MPC knockdown also reduced the abundance of several intermediates of the TCA cycle, and diminished α-ketoglutarate played a key role in attenuating HSC activation by suppressing hypoxia inducible factor-1α signaling. On high fat diets, mice with HSC-specific MPC deletion exhibited reduced circulating transaminases, numbers of HSC, and hepatic expression of markers of HSC activation and inflammation compared to wild-type mice. These data suggest that MPC inhibition modulates HSC metabolism to attenuate activation and illuminate mechanisms by which MPC inhibitors could prove therapeutically beneficial for treating MASH.
Figures
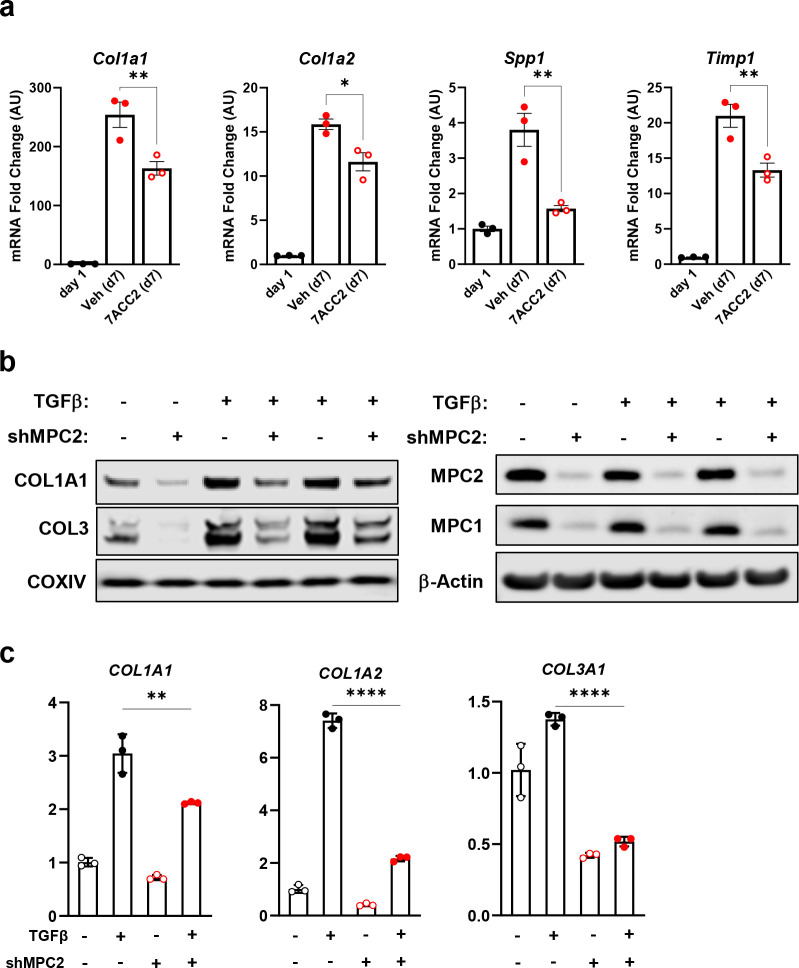
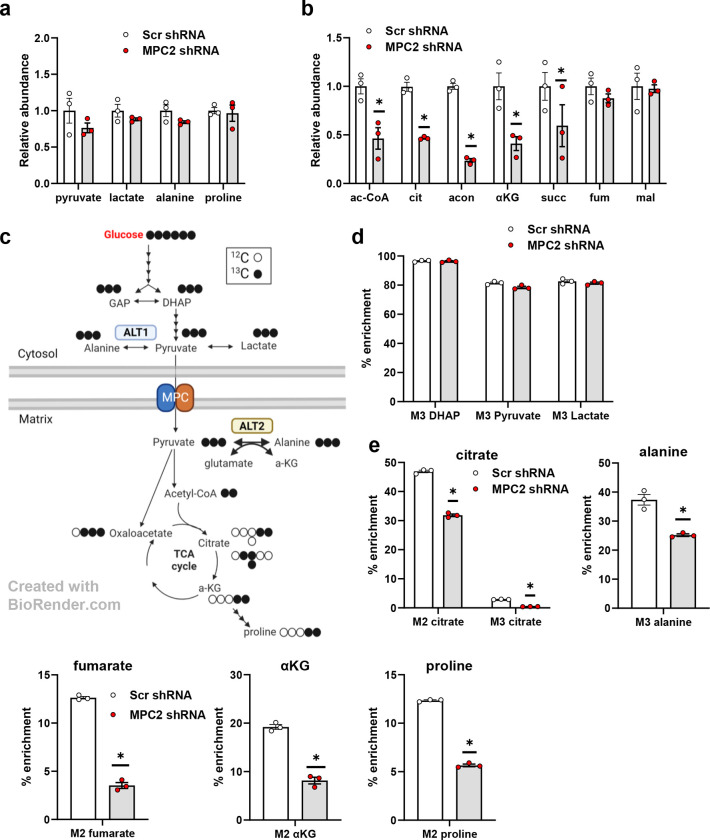
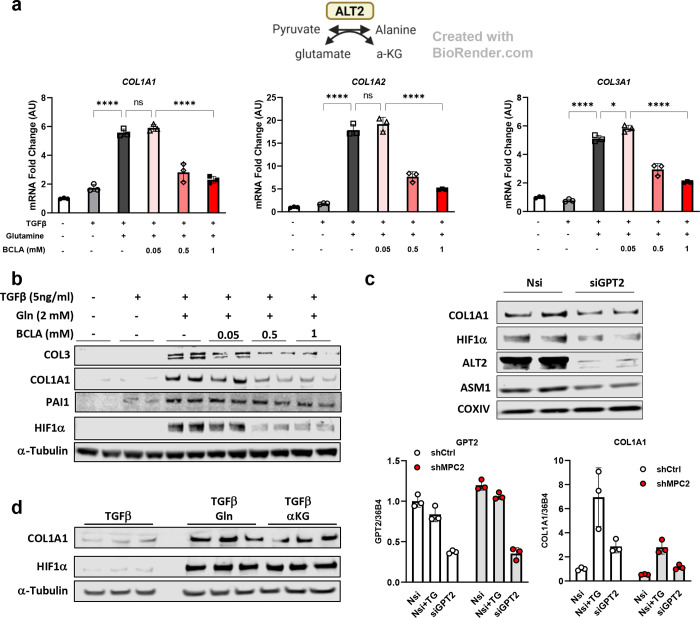
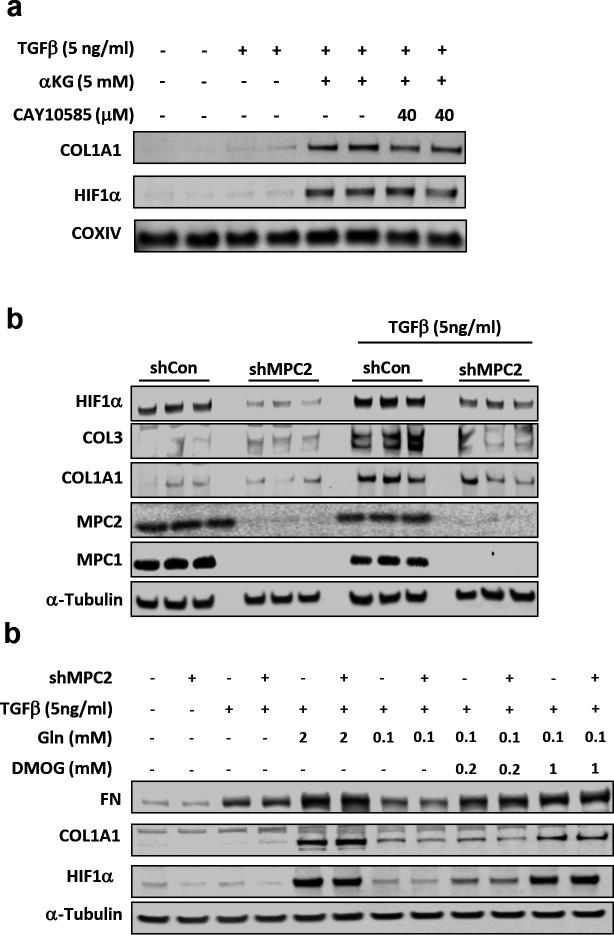
References
-
- Younossi Z.M., et al. , Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology, 2016. 64(1): p. 73–84. - PubMed
-
- Younossi Z., Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol., 2018. 15. - PubMed
-
- Taylor R.S., et al. , Association Between Fibrosis Stage and Outcomes of Patients With Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Gastroenterology, 2020. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources