

This is a preprint.
Reproducible evaluation of transposable element detectors with McClintock 2 guides accurate inference of Ty insertion patterns in yeast
- PMID: 36824955
- PMCID: PMC9948991
- DOI: 10.1101/2023.02.13.528343
Reproducible evaluation of transposable element detectors with McClintock 2 guides accurate inference of Ty insertion patterns in yeast
Update in
-
Reproducible evaluation of transposable element detectors with McClintock 2 guides accurate inference of Ty insertion patterns in yeast.Mob DNA. 2023 Jul 14;14(1):8. doi: 10.1186/s13100-023-00296-4. Mob DNA. 2023. PMID: 37452430 Free PMC article.
Abstract
Background: Many computational methods have been developed to detect non-reference transposable element (TE) insertions using short-read whole genome sequencing data. The diversity and complexity of such methods often present challenges to new users seeking to reproducibly install, execute, or evaluate multiple TE insertion detectors.
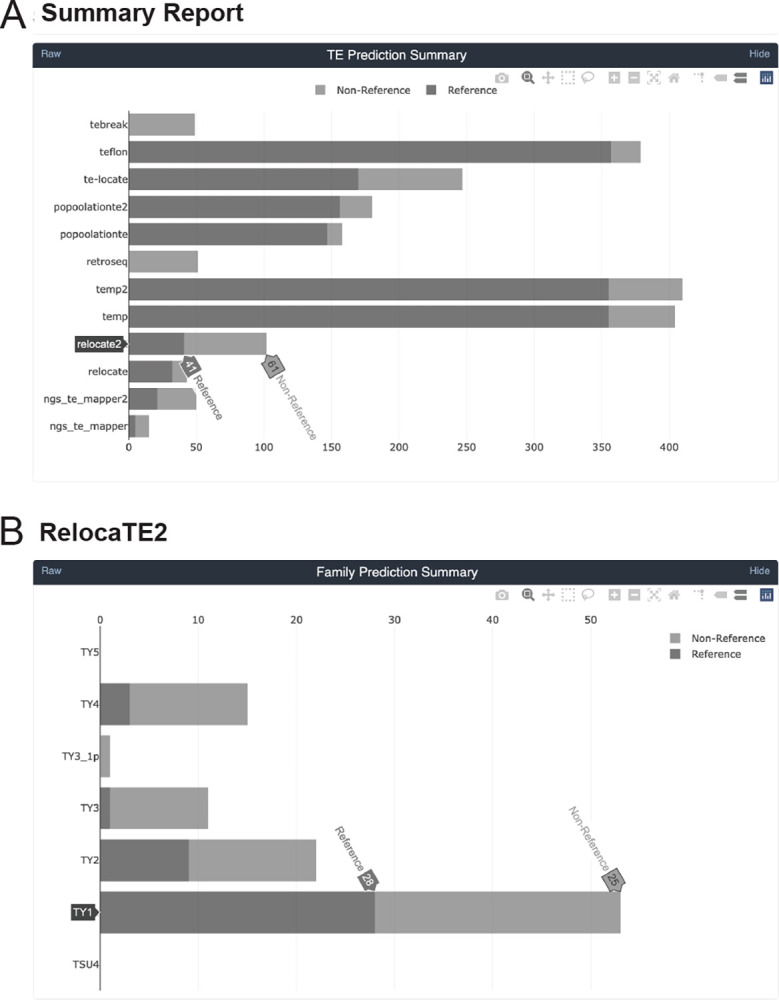
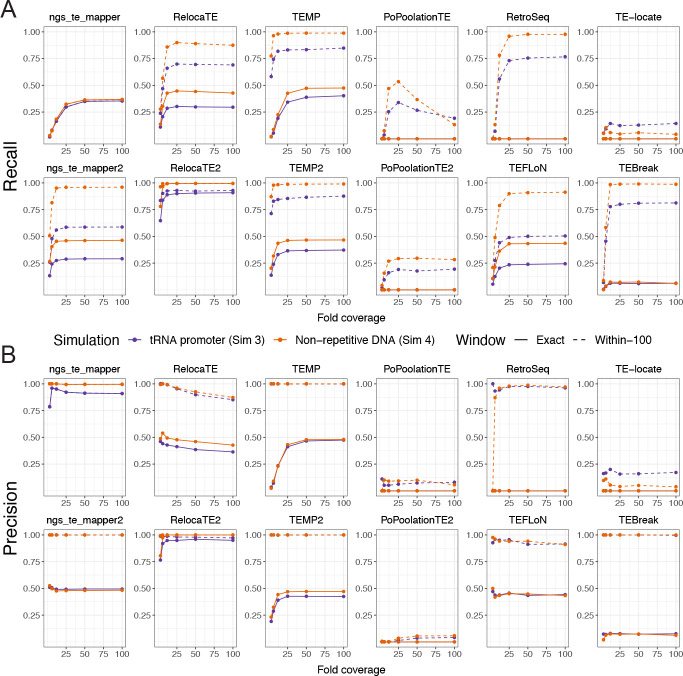
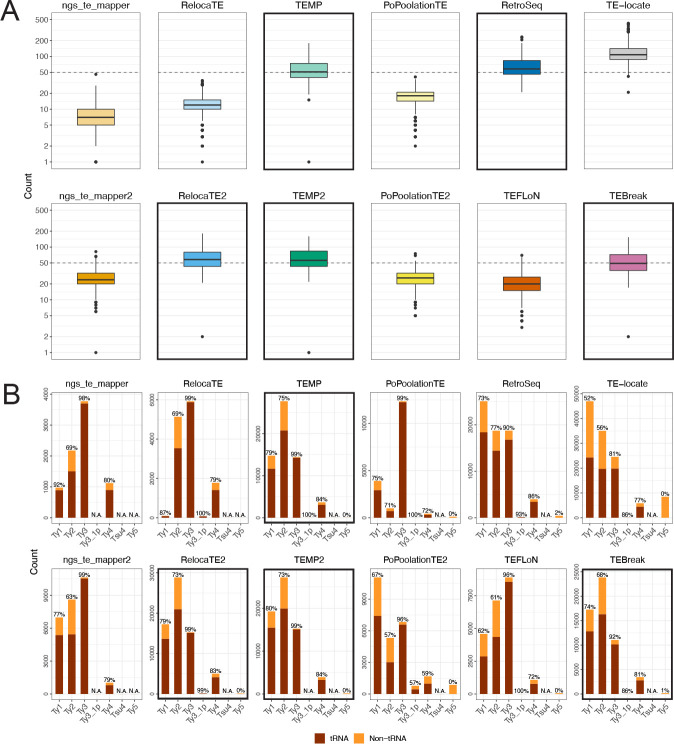
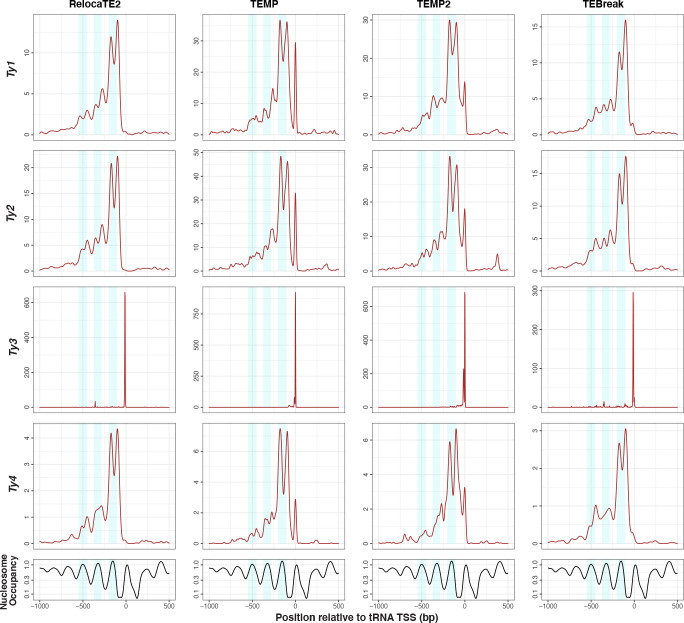
Results: We previously developed the McClintock meta-pipeline to facilitate the installation, execution, and evaluation of six first-generation short-read TE detectors. Here, we report a completely re-implemented version of McClintock written in Python using Snakemake and Conda that improves its installation, error handling, speed, stability, and extensibility. McClintock 2 now includes 12 short-read TE detectors, auxiliary pre-processing and analysis modules, interactive HTML reports, and a simulation framework to reproducibly evaluate the accuracy of component TE detectors. When applied to the model microbial eukaryote Saccharomyces cerevisiae, we find substantial variation in the ability of McClintock 2 components to identify the precise locations of non-reference TE insertions, with RelocaTE2 showing the highest recall and precision in simulated data. We find that RelocaTE2, TEMP, TEMP2 and TEBreak provide a consistent and biologically meaningful view of non-reference TE insertions in a species-wide panel of ∼1000 yeast genomes, as evaluated by coverage-based abundance estimates and expected patterns of tRNA promoter targeting. Finally, we show that best-in-class predictors for yeast have sufficient resolution to reveal a dyad pattern of integration in nucleosome-bound regions upstream of yeast tRNA genes for Ty1, Ty2, and Ty4, allowing us to extend knowledge about fine-scale target preferences first revealed experimentally for Ty1 to natural insertions and related copia-superfamily retrotransposons in yeast.
Conclusion: McClintock (https://github.com/bergmanlab/mcclintock/) provides a user-friendly pipeline for the identification of TEs in short-read WGS data using multiple TE detectors, which should benefit researchers studying TE insertion variation in a wide range of different organisms. Application of the improved McClintock system to simulated and empirical yeast genome data reveals best-in-class methods and novel biological insights for one of the most widely-studied model eukaryotes and provides a paradigm for evaluating and selecting non-reference TE detectors for other species.
Conflict of interest statement
Competing interests
The authors declare that they have no competing interests.
Figures

References
-
- Saha S., Bridges S., Magbanua Z.V., Peterson D.G.: Computational approaches and tools used in identification of dispersed repetitive DNA sequences. Tropical Plant Biol 1(1), 85–96 (2008). doi:10.1007/s12042-007-9007-5 - DOI
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources