

This is a preprint.
Pairtools: from sequencing data to chromosome contacts
- PMID: 36824968
- PMCID: PMC9949071
- DOI: 10.1101/2023.02.13.528389
Pairtools: from sequencing data to chromosome contacts
Update in
-
Pairtools: From sequencing data to chromosome contacts.PLoS Comput Biol. 2024 May 29;20(5):e1012164. doi: 10.1371/journal.pcbi.1012164. eCollection 2024 May. PLoS Comput Biol. 2024. PMID: 38809952 Free PMC article.
Abstract
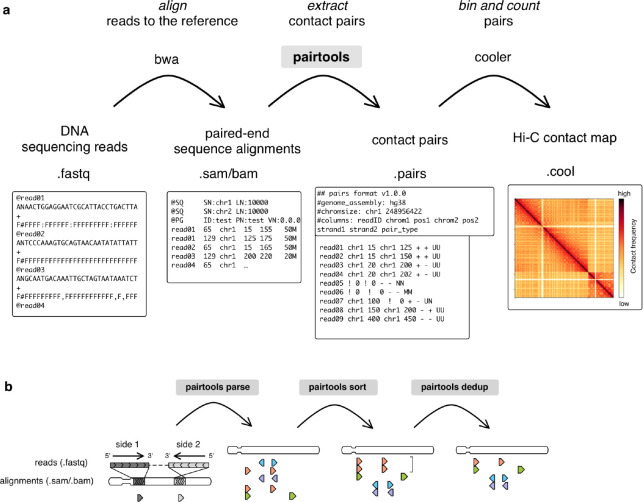
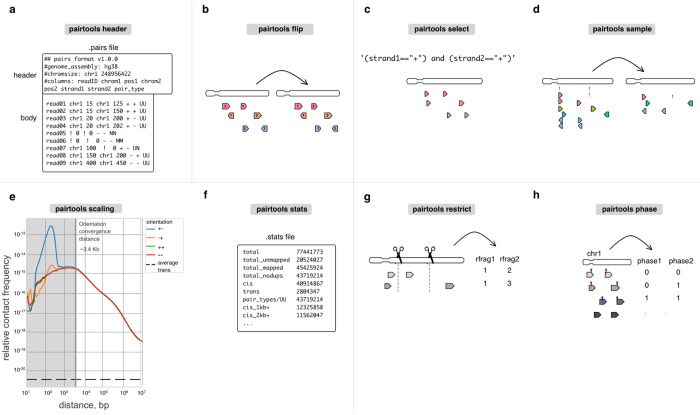
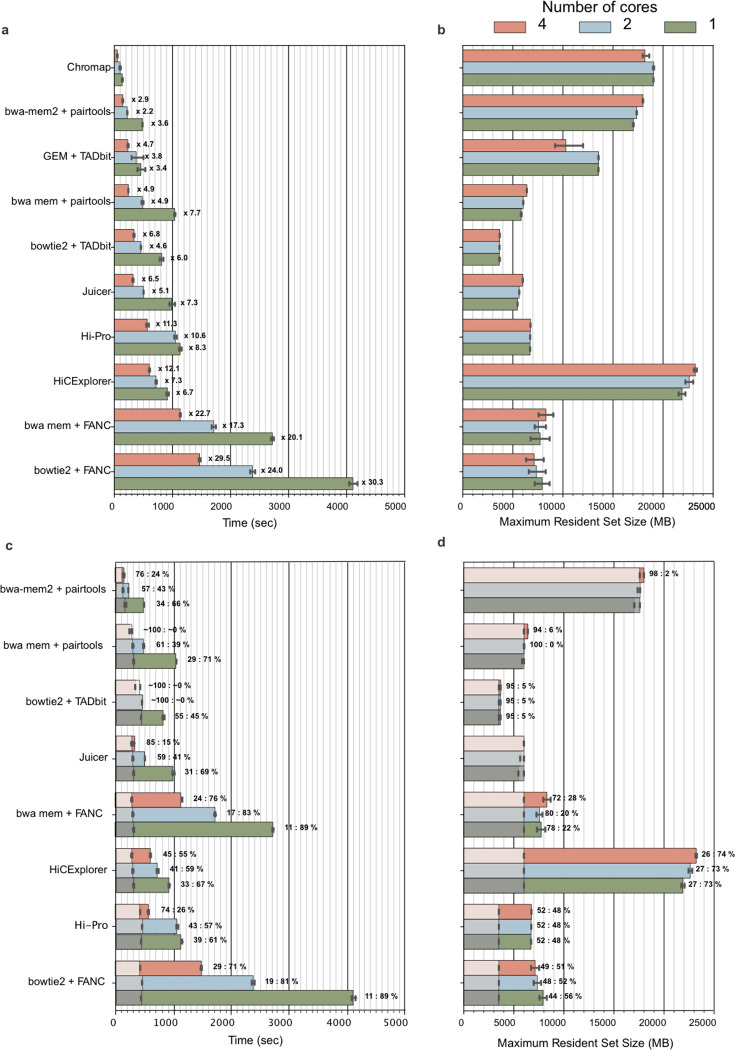
The field of 3D genome organization produces large amounts of sequencing data from Hi-C and a rapidly-expanding set of other chromosome conformation protocols (3C+). Massive and heterogeneous 3C+ data require high-performance and flexible processing of sequenced reads into contact pairs. To meet these challenges, we present pairtools - a flexible suite of tools for contact extraction from sequencing data. Pairtools provides modular command-line interface (CLI) tools that can be flexibly chained into data processing pipelines. Pairtools provides both crucial core tools as well as auxiliary tools for building feature-rich 3C+ pipelines, including contact pair manipulation, filtration, and quality control. Benchmarking pairtools against popular 3C+ data pipelines shows advantages of pairtools for high-performance and flexible 3C+ analysis. Finally, pairtools provides protocol-specific tools for multi-way contacts, haplotype-resolved contacts, and single-cell Hi-C. The combination of CLI tools and tight integration with Python data analysis libraries makes pairtools a versatile foundation for a broad range of 3C+ pipelines.
Figures

References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous