

Variant-specific deleterious mutations in the SARS-CoV-2 genome reveal immune responses and potentials for prophylactic vaccine development
- PMID: 36825152
- PMCID: PMC9941545
- DOI: 10.3389/fphar.2023.1090717
Variant-specific deleterious mutations in the SARS-CoV-2 genome reveal immune responses and potentials for prophylactic vaccine development
Abstract
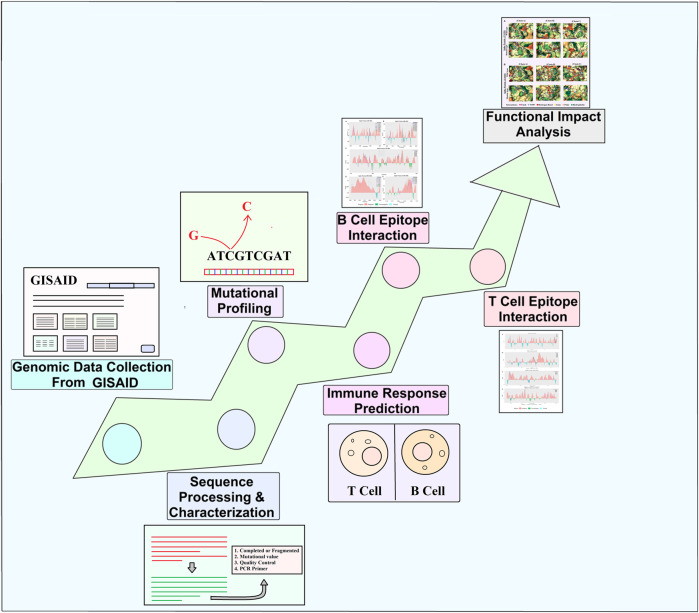
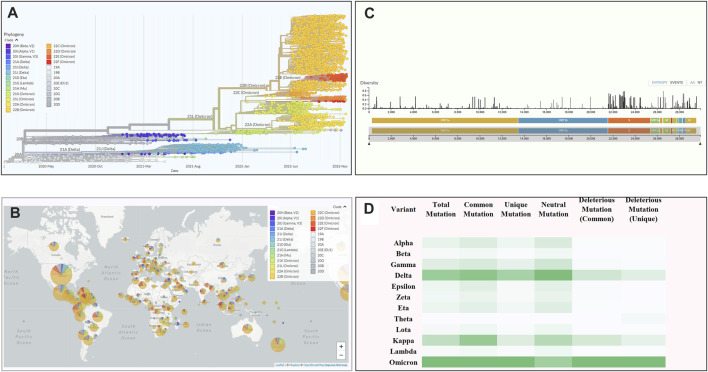
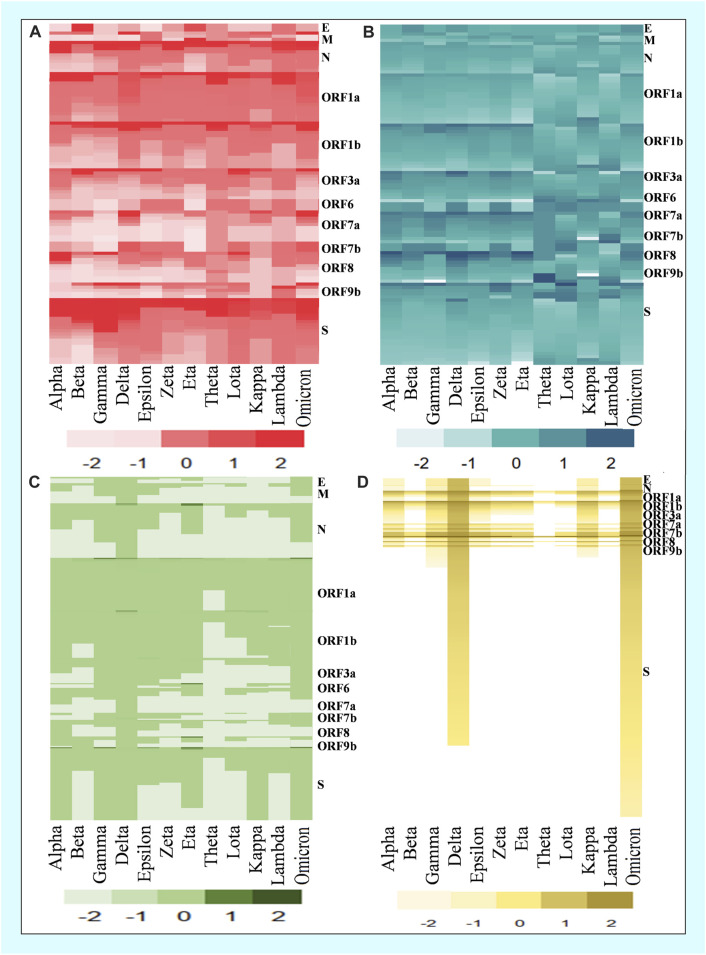
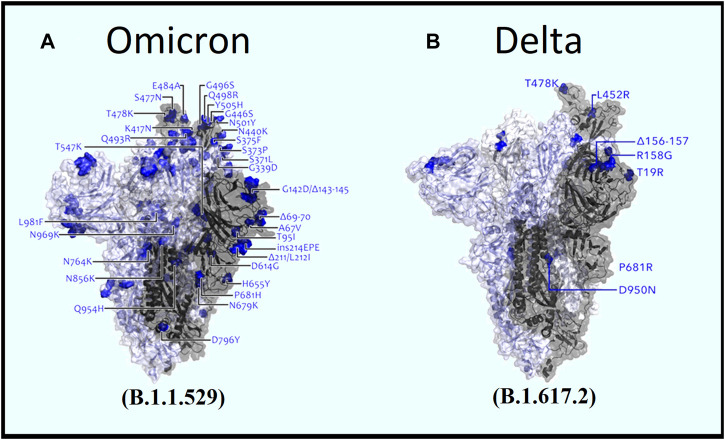
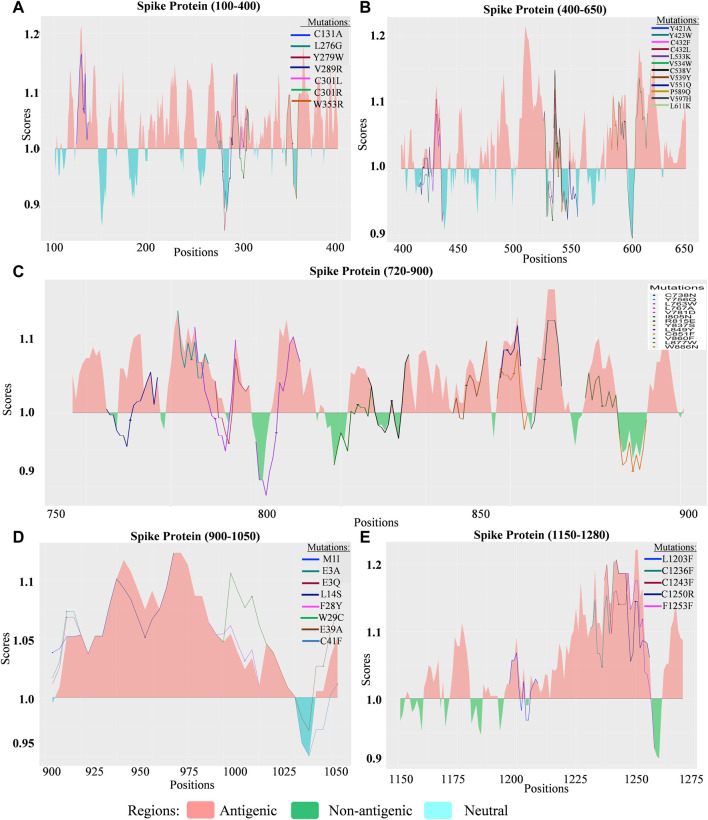
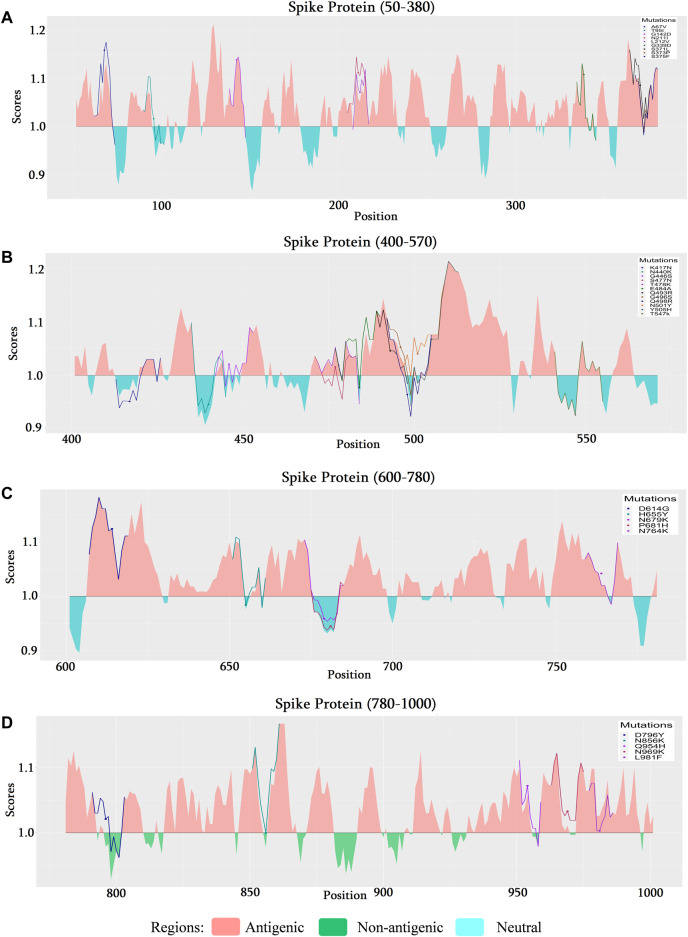
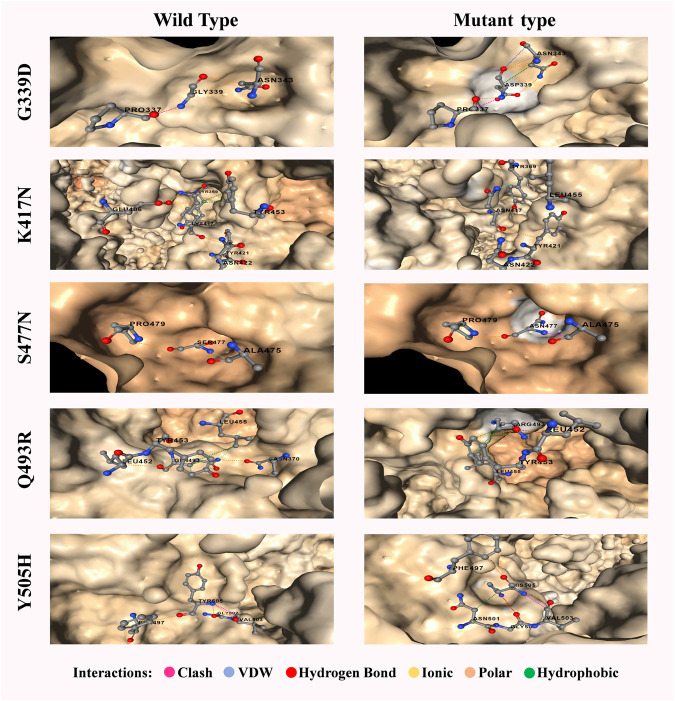
Introduction: Coronavirus disease 2019 (COVID-19), caused by SARS-CoV-2, has had a disastrous effect worldwide during the previous three years due to widespread infections with SARS-CoV-2 and its emerging variations. More than 674 million confirmed cases and over 6.7 million deaths have been attributed to successive waves of SARS-CoV-2 infections as of 29th January 2023. Similar to other RNA viruses, SARS-CoV-2 is more susceptible to genetic evolution and spontaneous mutations over time, resulting in the continual emergence of variants with distinct characteristics. Spontaneous mutations of SARS-CoV-2 variants increase its transmissibility, virulence, and disease severity and diminish the efficacy of therapeutics and vaccines, resulting in vaccine-breakthrough infections and re-infection, leading to high mortality and morbidity rates. Materials and methods: In this study, we evaluated 10,531 whole genome sequences of all reported variants globally through a computational approach to assess the spread and emergence of the mutations in the SARS-CoV-2 genome. The available data sources of NextCladeCLI 2.3.0 (https://clades.nextstrain.org/) and NextStrain (https://nextstrain.org/) were searched for tracking SARS-CoV-2 mutations, analysed using the PROVEAN, Polyphen-2, and Predict SNP mutational analysis tools and validated by Machine Learning models. Result: Compared to the Wuhan-Hu-1 reference strain NC 045512.2, genome-wide annotations showed 16,954 mutations in the SARS-CoV-2 genome. We determined that the Omicron variant had 6,307 mutations (retrieved sequence:1947), including 67.8% unique mutations, more than any other variant evaluated in this study. The spike protein of the Omicron variant harboured 876 mutations, including 443 deleterious mutations. Among these deleterious mutations, 187 were common and 256 were unique non-synonymous mutations. In contrast, after analysing 1,884 sequences of the Delta variant, we discovered 4,468 mutations, of which 66% were unique, and not previously reported in other variants. Mutations affecting spike proteins are mostly found in RBD regions for Omicron, whereas most of the Delta variant mutations drawn to focus on amino acid regions ranging from 911 to 924 in the context of epitope prediction (B cell & T cell) and mutational stability impact analysis protruding that Omicron is more transmissible. Discussion: The pathogenesis of the Omicron variant could be prevented if the deleterious and persistent unique immunosuppressive mutations can be targeted for vaccination or small-molecule inhibitor designing. Thus, our findings will help researchers monitor and track the continuously evolving nature of SARS-CoV-2 strains, the associated genetic variants, and their implications for developing effective control and prophylaxis strategies.
Keywords: COVID-19; SARS-CoV-2; deleterious mutation; delta variant; immune response; omicron variant; unique mutation; vaccine designing.
Copyright © 2023 Islam, Shahi, Marzan, Amin, Hasan, Hoque, Ghosh, Barua, Khan, Dhama, Chakraborty, Bhattacharya and Wei.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures

References
-
- Ahmed F., Islam M. A., Kumar M., Hossain M., Bhattacharya P., Islam M. T., et al. (2021). First detection of SARS-CoV-2 genetic material in the vicinity of COVID-19 isolation Centre in Bangladesh: Variation along the sewer network. Sci. Total Environ. 776, 145724. 10.1016/j.scitotenv.2021.145724 - DOI - PMC - PubMed
-
- Aksamentov I., Roemer C., Hodcroft E., Neher R. (2021). Nextclade: Clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 6 (67), 3773. 10.21105/joss.03773 - DOI
-
- Aleem A., Akbar Samad A. B., Slenker A. K. (2022). Emerging variants of SARS-CoV-2 and novel therapeutics against coronavirus (COVID-19). reasure Island: StatPearls. - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous