

The synthetic TRPML1 agonist ML-SA1 rescues Alzheimer-related alterations of the endosomal-autophagic-lysosomal system
- PMID: 36825945
- PMCID: PMC10112969
- DOI: 10.1242/jcs.259875
The synthetic TRPML1 agonist ML-SA1 rescues Alzheimer-related alterations of the endosomal-autophagic-lysosomal system
Abstract
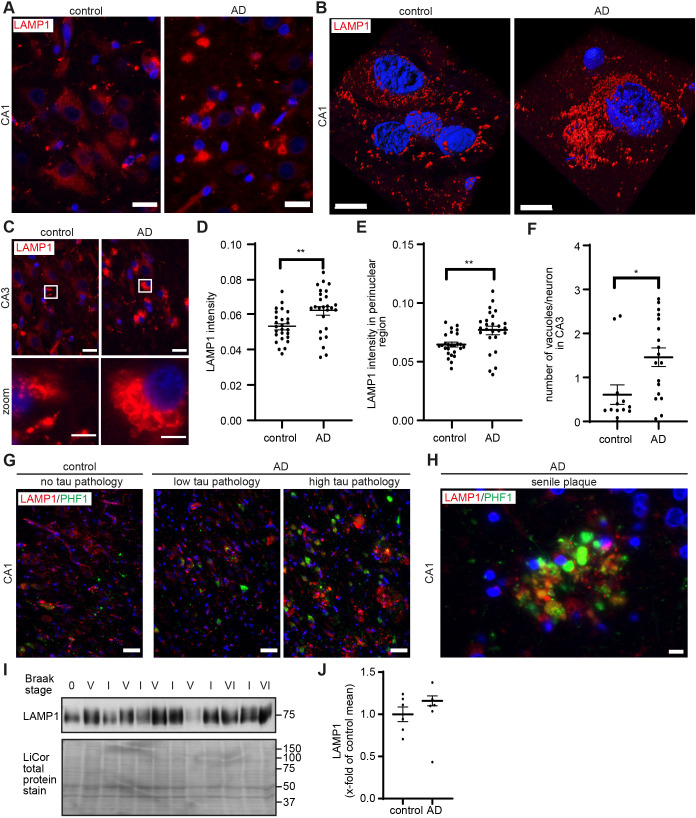
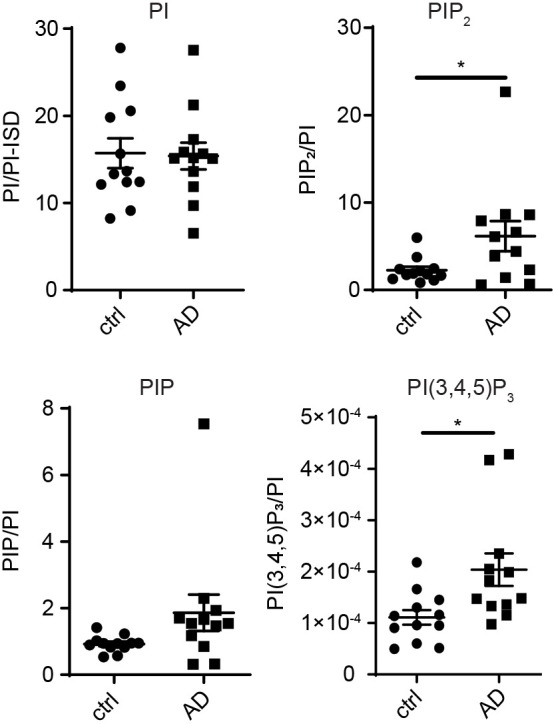
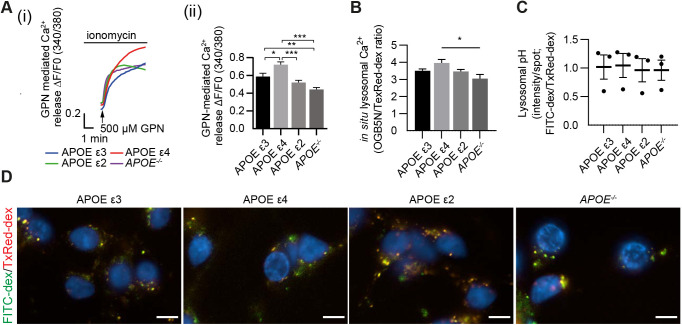
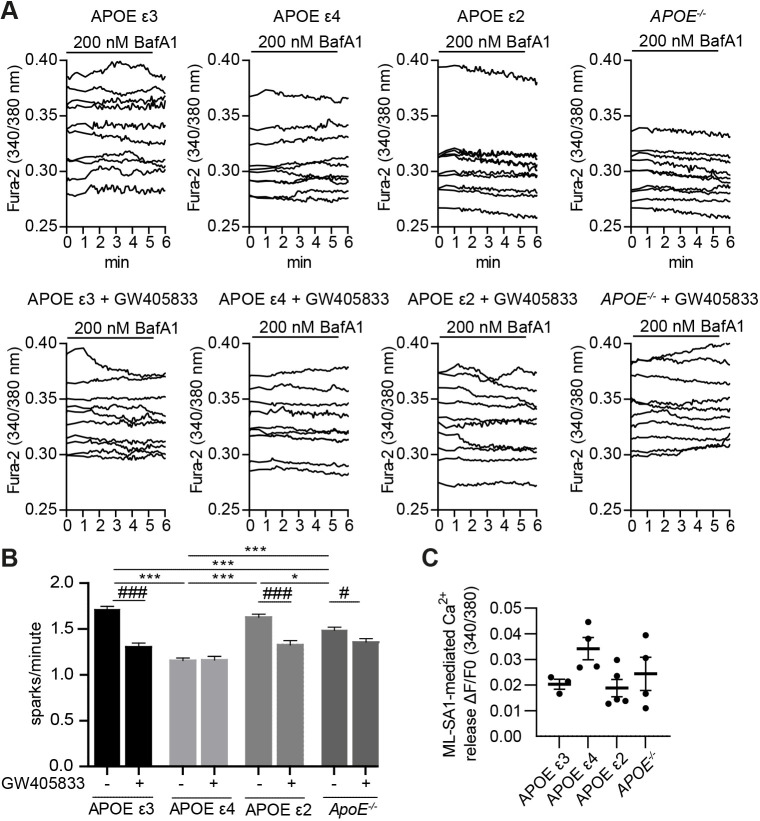
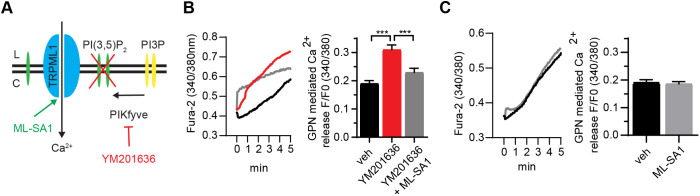
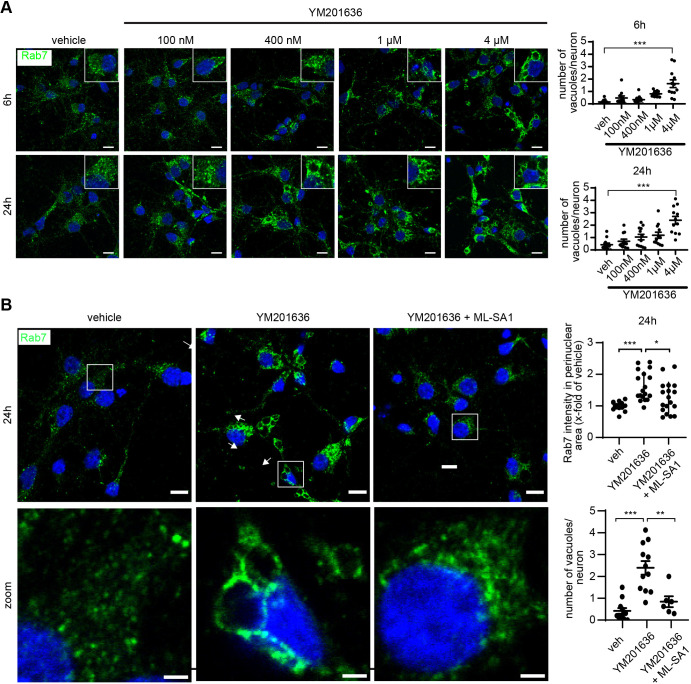
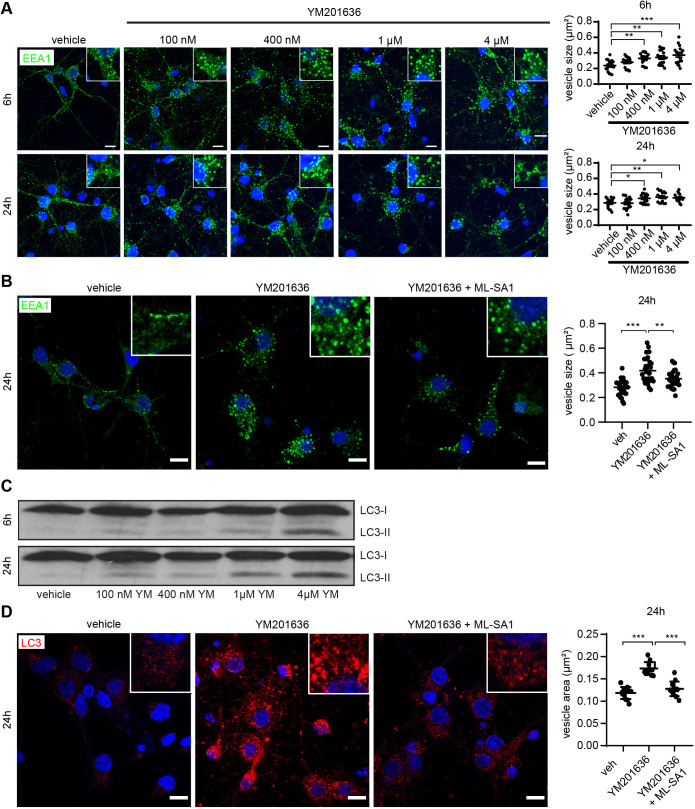
Abnormalities in the endosomal-autophagic-lysosomal (EAL) system are an early event in Alzheimer's disease (AD) pathogenesis. However, the mechanisms underlying these abnormalities are unclear. The transient receptor potential channel mucolipin 1(TRPML1, also known as MCOLN1), a vital endosomal-lysosomal Ca2+ channel whose loss of function leads to neurodegeneration, has not been investigated with respect to EAL pathogenesis in late-onset AD (LOAD). Here, we identify pathological hallmarks of TRPML1 dysregulation in LOAD neurons, including increased perinuclear clustering and vacuolation of endolysosomes. We reveal that induced pluripotent stem cell (iPSC)-derived human cortical neurons expressing APOE ε4, the strongest genetic risk factor for LOAD, have significantly diminished TRPML1-induced endolysosomal Ca2+ release. Furthermore, we found that blocking TRPML1 function in primary neurons by depleting the TRPML1 agonist PI(3,5)P2 via PIKfyve inhibition, recreated multiple features of EAL neuropathology evident in LOAD. This included increased endolysosomal Ca2+ content, enlargement and perinuclear clustering of endolysosomes, autophagic vesicle accumulation and early endosomal enlargement. Strikingly, these AD-like neuronal EAL defects were rescued by TRPML1 reactivation using its synthetic agonist ML-SA1. These findings implicate defects in TRPML1 in LOAD EAL pathogenesis and present TRPML1 as a potential therapeutic target.
Keywords: APOE; Alzheimer's disease; Ca2+; PIKfyve; Phosphoinositides; TRPML1.
© 2023. Published by The Company of Biologists Ltd.
Conflict of interest statement
Competing interests The authors declare no competing or financial interests.
Figures
References
-
- Akwa, Y., Di Malta, C., Zallo, F., Gondard, E., Lunati, A., Diaz-De-Grenu, L. Z., Zampelli, A., Boiret, A., Santamaria, S., Martinez-Preciado, M.et al. (2023). Stimulation of synaptic activity promotes TFEB-mediated clearance of pathological MAPT/Tau in cellular and mouse models of tauopathies. Autophagy 19, 660-677. 10.1080/15548627.2022.2095791 - DOI - PMC - PubMed
-
- Bae, M., Patel, N., Xu, H., Lee, M., Tominaga-Yamanaka, K., Nath, A., Geiger, J., Gorospe, M., Mattson, M. P. and Haughey, N. J. (2014). Activation of TRPML1 clears intraneuronal Aβ in preclinical models of HIV infection. J. Neurosci. 34, 11485-11503. 10.1523/JNEUROSCI.0210-14.2014 - DOI - PMC - PubMed
-
- Bargal, R., Avidan, N., Olender, T., Ben Asher, E., Zeigler, M., Raas-Rothschild, A., Frumkin, A., Ben-Yoseph, O., Friedlender, Y., Lancet, D.et al. (2001). Mucolipidosis type IV: novel MCOLN1 mutations in Jewish and non-Jewish patients and the frequency of the disease in the Ashkenazi Jewish population. Hum. Mutat. 17, 397-402. 10.1002/humu.1115 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- BBS/E/B/000C0415/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- MC_EX_MR/N50192X/1/MRC_/Medical Research Council/United Kingdom
- BB/J004456/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- SFI15/COEN/3037/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
