

The Homogeneous Azorean Machado-Joseph Disease Cohort: Characterization and Contributions to Advances in Research
- PMID: 36830784
- PMCID: PMC9953730
- DOI: 10.3390/biomedicines11020247
The Homogeneous Azorean Machado-Joseph Disease Cohort: Characterization and Contributions to Advances in Research
Abstract
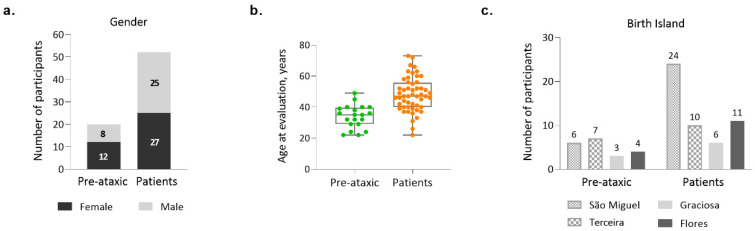
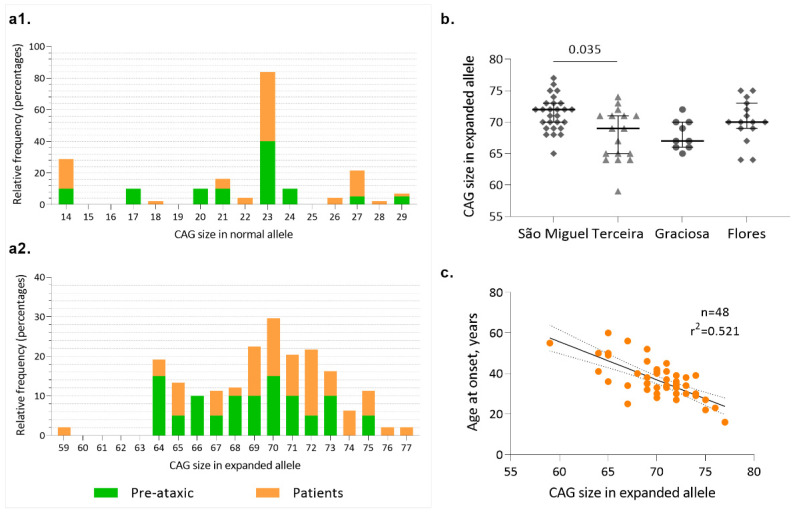
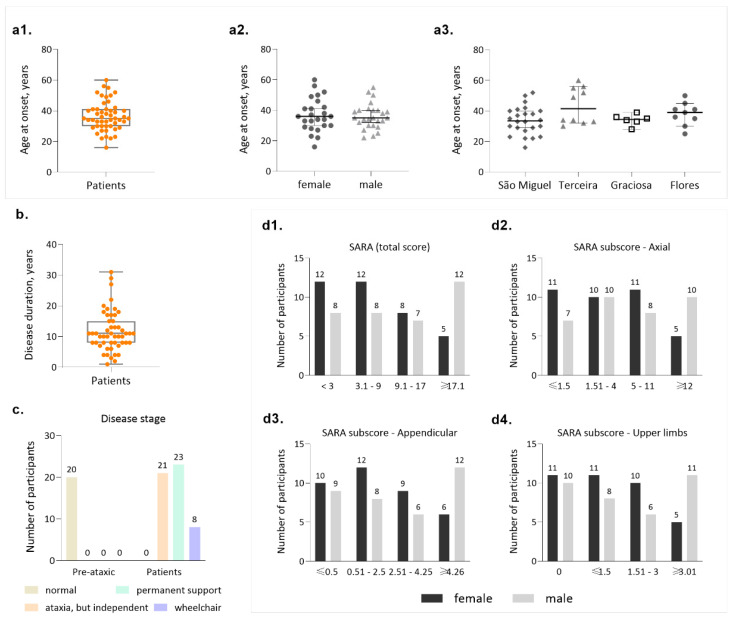
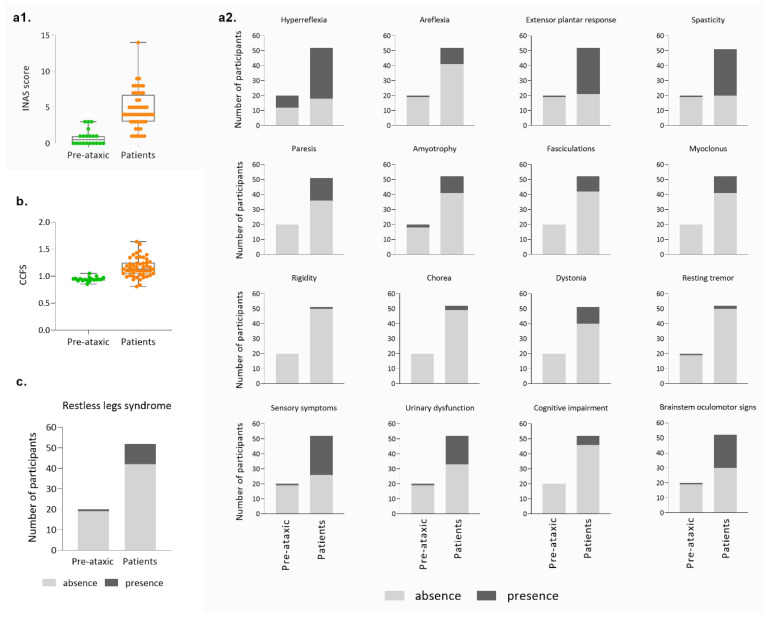
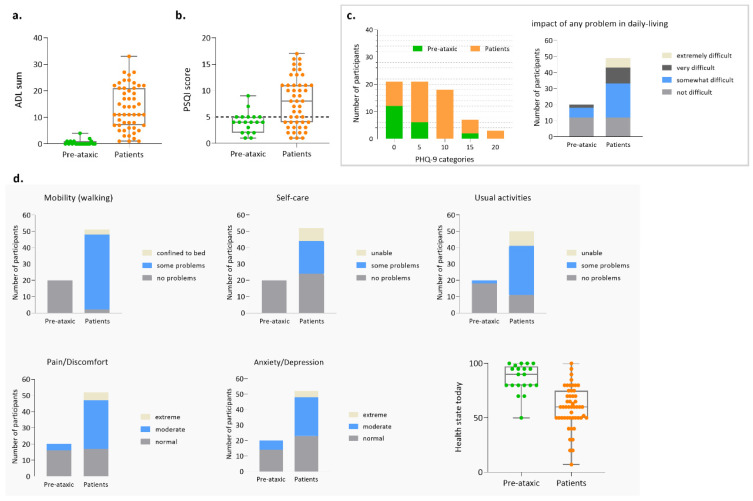
Machado-Joseph disease (MJD)/spinocerebellar ataxia type 3 (SCA3) is the most common autosomal dominant ataxia worldwide. MJD is characterized by late-onset progressive cerebellar ataxia associated with variable clinical findings, including pyramidal signs and a dystonic-rigid extrapyramidal syndrome. In the Portuguese archipelago of the Azores, the worldwide population cluster for this disorder (prevalence of 39 in 100,000 inhabitants), a cohort of MJD mutation carriers belonging to extensively studied pedigrees has been followed since the late 1990s. Studies of the homogeneous Azorean MJD cohort have been contributing crucial information to the natural history of this disease as well as allowing the identification of novel molecular biomarkers. Moreover, as interventional studies for this globally rare and yet untreatable disease are emerging, this cohort should be even more important for the recruitment of trial participants. In this paper, we profile the Azorean cohort of MJD carriers, constituted at baseline by 20 pre-ataxic carriers and 52 patients, which currently integrates the European spinocerebellar ataxia type 3/Machado-Joseph disease Initiative (ESMI), a large European longitudinal MJD cohort. Moreover, we summarize the main studies based on this cohort and highlight the contributions made to advances in MJD research. Knowledge of the profile of the Azorean MJD cohort is not only important in the context of emergent interventional trials but is also pertinent for the implementation of adequate interventional measures, constituting relevant information for Lay Associations and providing data to guide healthcare decision makers.
Keywords: European Spinocerebellar Ataxia type 3/Machado-Joseph disease Initiative (ESMI); MJD; SCA3; clinical trials; homogeneous cohorts; polyglutamine disorders; spinocerebellar ataxia type 3.
Conflict of interest statement
The authors declare that they have no competing interest.
Figures

References
-
- de Araujo M., Raposo M., Kazachkova N., Vasconcelos J., Kay T., Lima M. Trends in the Epidemiology of Spinocerebellar Ataxia Type 3 / Machado-Joseph Disease in the Azores Islands, Portugal. JSM Brain Sci. 2016;1:1001.
-
- Paulson H., Shakkottai V. Spinocerebellar Ataxia Type 3. In: Adam M.P., Everman D.B., Mirzaa G.M., Pagon R.A., Wallace S.E., Bean L.J.H., Gripp K.W., Amemiya A., editors. GeneReviews®. University of Washington; Seattle, WA, USA: 1998. pp. 1993–2022. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
