

PD-1/PD-L1 and DNA Damage Response in Cancer
- PMID: 36831197
- PMCID: PMC9954559
- DOI: 10.3390/cells12040530
PD-1/PD-L1 and DNA Damage Response in Cancer
Abstract
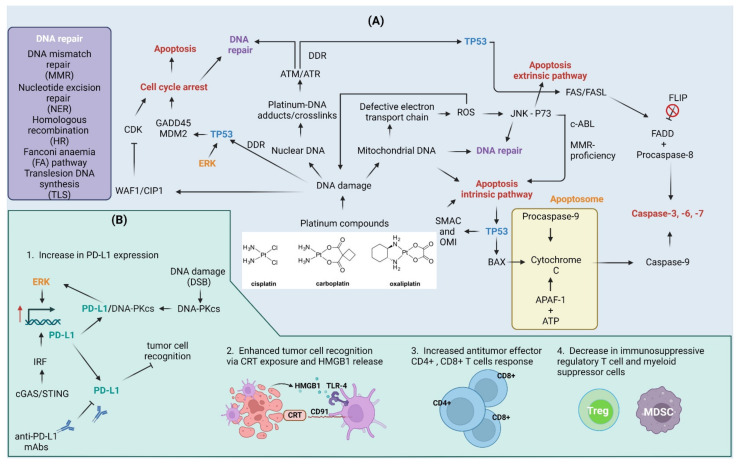
The application of immunotherapy for cancer treatment is rapidly becoming more widespread. Immunotherapeutic agents are frequently combined with various types of treatments to obtain a more durable antitumor clinical response in patients who have developed resistance to monotherapy. Chemotherapeutic drugs that induce DNA damage and trigger DNA damage response (DDR) frequently induce an increase in the expression of the programmed death ligand-1 (PD-L1) that can be employed by cancer cells to avoid immune surveillance. PD-L1 exposed on cancer cells can in turn be targeted to re-establish the immune-reactive tumor microenvironment, which ultimately increases the tumor's susceptibility to combined therapies. Here we review the recent advances in how the DDR regulates PD-L1 expression and point out the effect of etoposide, irinotecan, and platinum compounds on the anti-tumor immune response.
Keywords: DNA damage response; cytotoxic drugs; immunotherapy; programmed death ligand-1 (PD-L1).
Conflict of interest statement
The authors declare no conflict of interest.
Figures




References
-
- Gardai S.J., McPhillips K.A., Frasch S.C., Janssen W.J., Starefeldt A., Murphy-Ullrich J.E., Bratton D.L., Oldenborg P.-A., Michalak M., Henson P.M. Cell-Surface Calreticulin Initiates Clearance of Viable or Apoptotic Cells through Trans-Activation of LRP on the Phagocyte. Cell. 2005;123:321–334. doi: 10.1016/j.cell.2005.08.032. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
