

Doxorubicin-An Agent with Multiple Mechanisms of Anticancer Activity
- PMID: 36831326
- PMCID: PMC9954613
- DOI: 10.3390/cells12040659
Doxorubicin-An Agent with Multiple Mechanisms of Anticancer Activity
Abstract
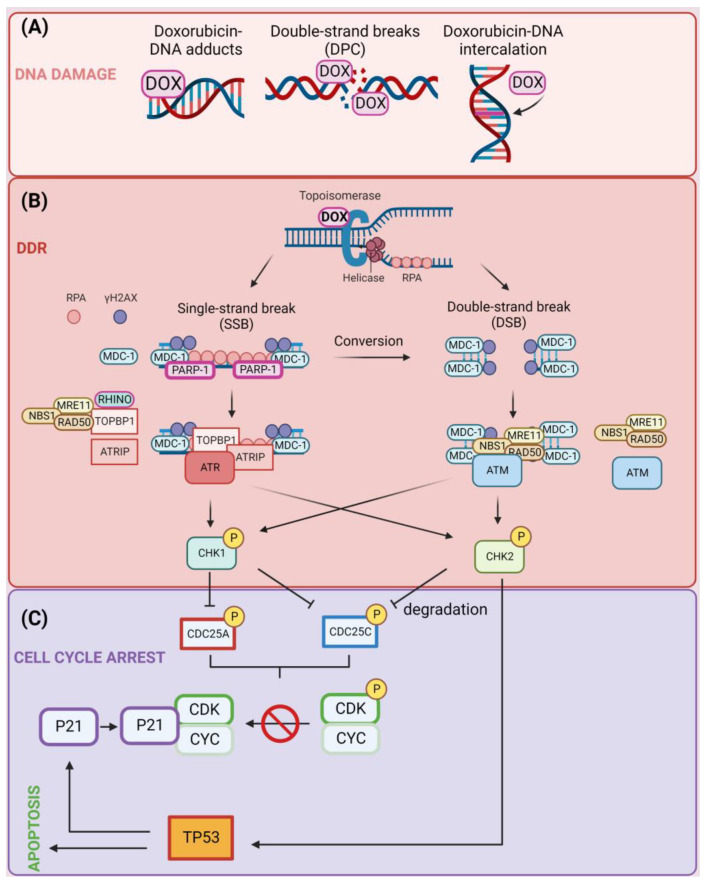
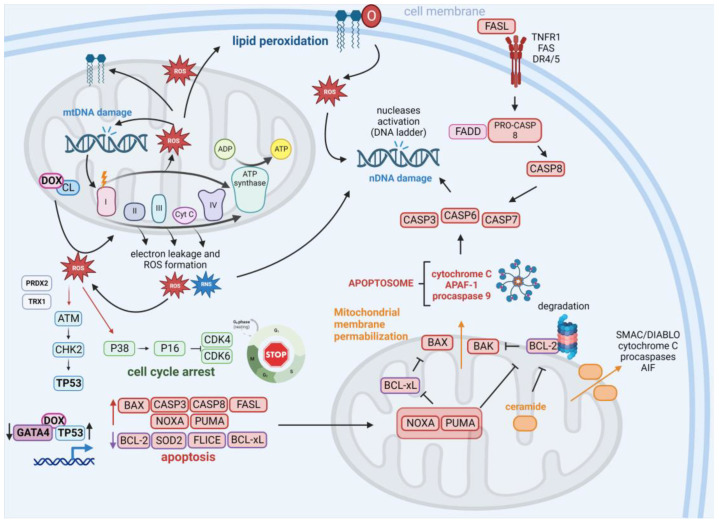
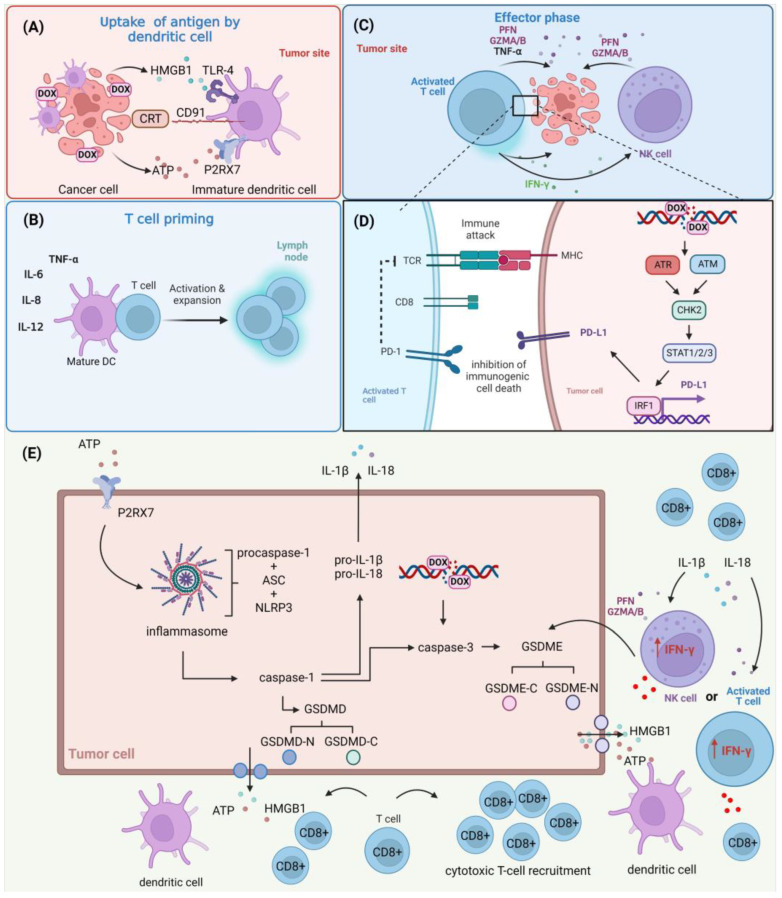
Doxorubicin (DOX) constitutes the major constituent of anti-cancer treatment regimens currently in clinical use. However, the precise mechanisms of DOX's action are not fully understood. Emerging evidence points to the pleiotropic anticancer activity of DOX, including its contribution to DNA damage, reactive oxygen species (ROS) production, apoptosis, senescence, autophagy, ferroptosis, and pyroptosis induction, as well as its immunomodulatory role. This review aims to collect information on the anticancer mechanisms of DOX as well as its influence on anti-tumor immune response, providing a rationale behind the importance of DOX in modern cancer therapy.
Keywords: DNA damage response (DDR); adriamycin; apoptosis; doxorubicin; immunotherapy.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
