

Spondyloocular Syndrome: A Report of an Additional Family and Phenotypic Spectrum Delineation
- PMID: 36833424
- PMCID: PMC9957273
- DOI: 10.3390/genes14020497
Spondyloocular Syndrome: A Report of an Additional Family and Phenotypic Spectrum Delineation
Abstract
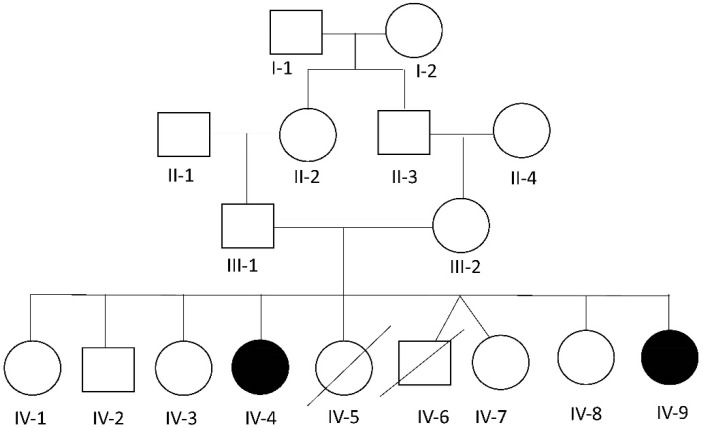
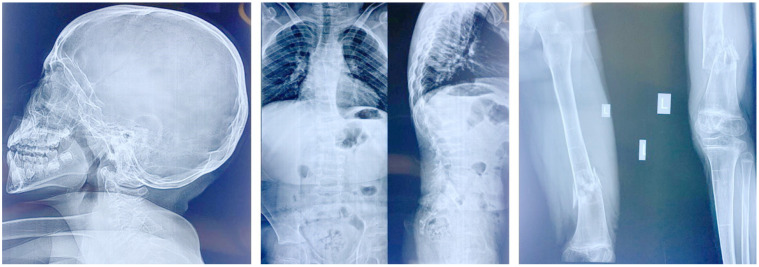
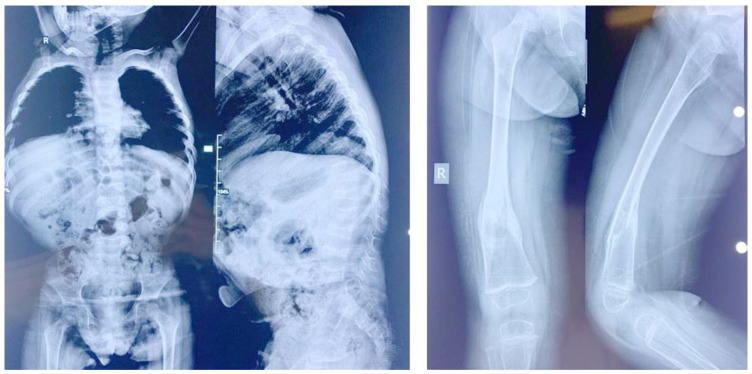
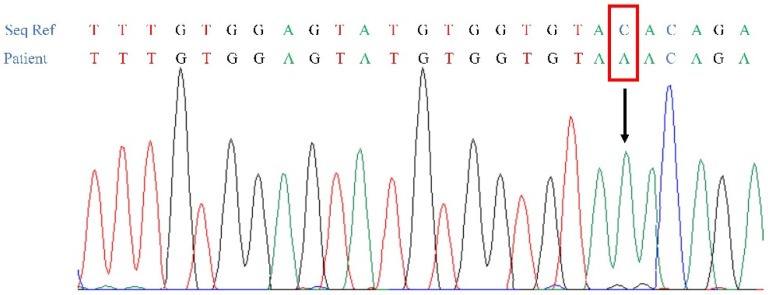
Spondyloocular syndrome (SOS, OMIM # 605822) is a rare genetic disorder characterized by osseous and ocular manifestations, including generalized osteoporosis, multiple long bones fractures, platyspondyly, dense cataracts and retinal detachment, and dysmorphic facial features, with or without short stature, cardiopathy, hearing impairment, and intellectual disability. Biallelic mutations in the XYLT2 gene (OMIM * 608125), encoding the xylosyltransferase II, were shown to be responsible for this disease. To date, 22 cases with SOS have been described, with varying clinical presentations and a yet-to-be-established genotypic-phenotypic correlation. Two patients from a consanguineous Lebanese family that presented with SOS were included in this study. Whole exome sequencing revealed a novel homozygous nonsense mutation in XYLT2 (p.Tyr414*) in these patients. We review all previously reported cases with SOS, describe the second nonsense mutation in XYLT2, and contribute to a better delineation of the phenotypic spectrum of the disease.
Keywords: Lebanon; XYLT2; ocular disorders; skeletal disorders; spondyloocular syndrome; whole exome sequencing.
Conflict of interest statement
Authors declare that no conflict of interests exist.
Figures
References
-
- Schmidt H., Rudolph G., Hergersberg M., Schneider K., Moradi S., Meitinger T. Retinal detachment and cataract, facial dysmorphism, generalized osteoporosis, immobile spine and platyspondyly in a consanguinous kindred—A possible new syndrome: A possible new spondylo-ocular syndrome. Clin. Genet. 2001;59:99–105. doi: 10.1034/j.1399-0004.2001.590206.x. - DOI - PubMed
-
- Munns C.F., Fahiminiya S., Poudel N., Munteanu M.C., Majewski J., Sillence D.O., Metcalf J.P., Biggin A., Glorieux F., Fassier F., et al. Homozygosity for Frameshift Mutations in XYLT2 Result in a Spondylo-Ocular Syndrome with Bone Fragility, Cataracts, and Hearing Defects. Am. J. Hum. Genet. 2015;96:971–978. doi: 10.1016/j.ajhg.2015.04.017. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
