

Vorinostat Improves Myotonic Dystrophy Type 1 Splicing Abnormalities in DM1 Muscle Cell Lines and Skeletal Muscle from a DM1 Mouse Model
- PMID: 36835205
- PMCID: PMC9964082
- DOI: 10.3390/ijms24043794
Vorinostat Improves Myotonic Dystrophy Type 1 Splicing Abnormalities in DM1 Muscle Cell Lines and Skeletal Muscle from a DM1 Mouse Model
Abstract
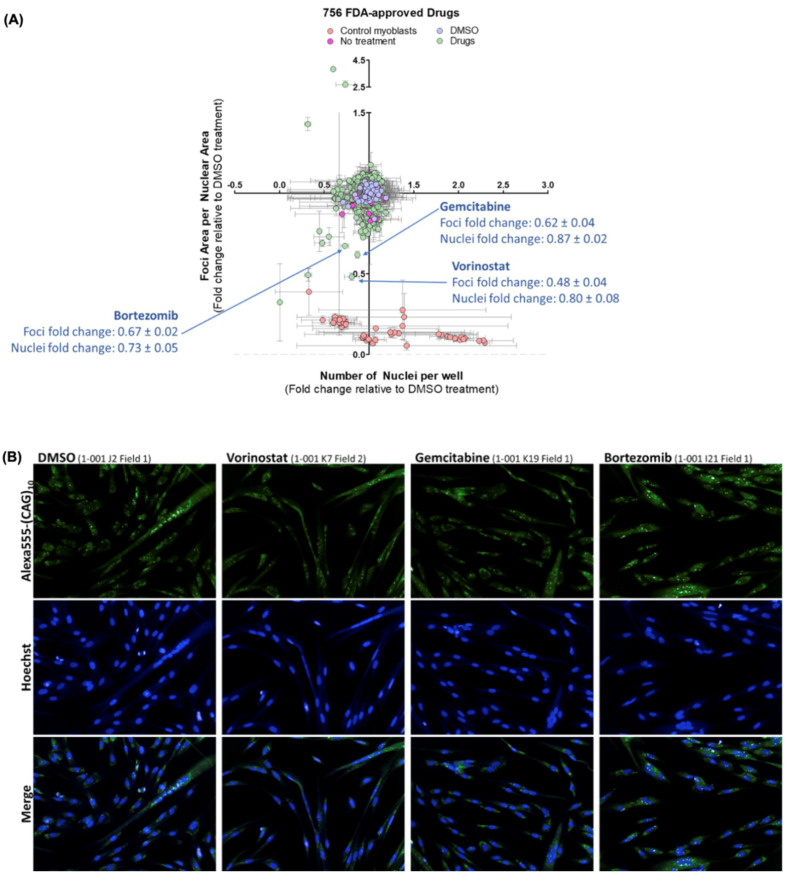
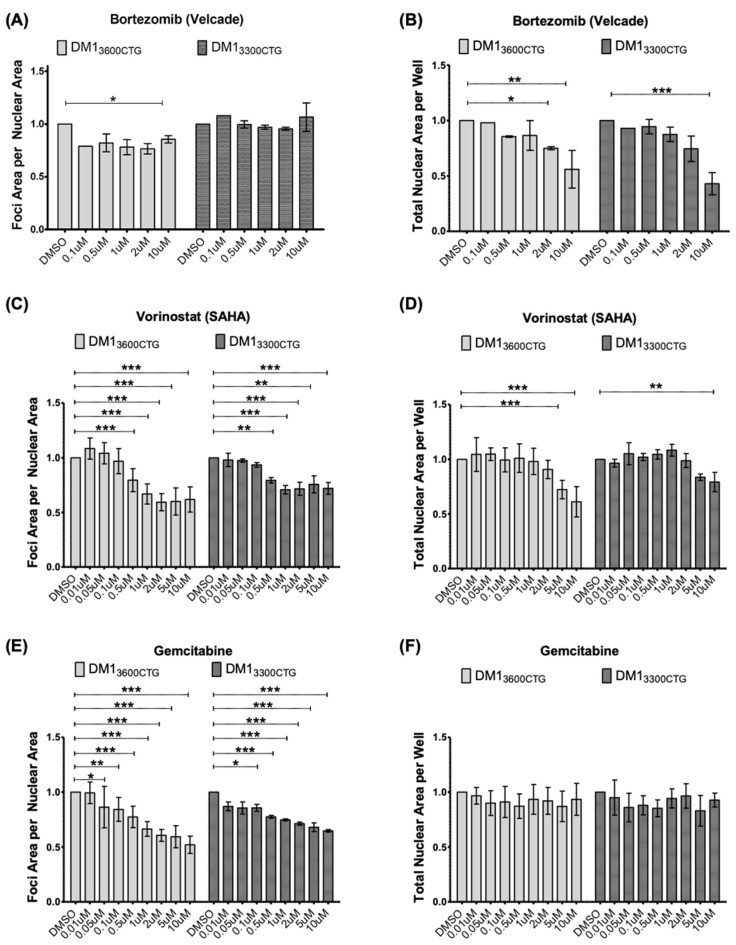
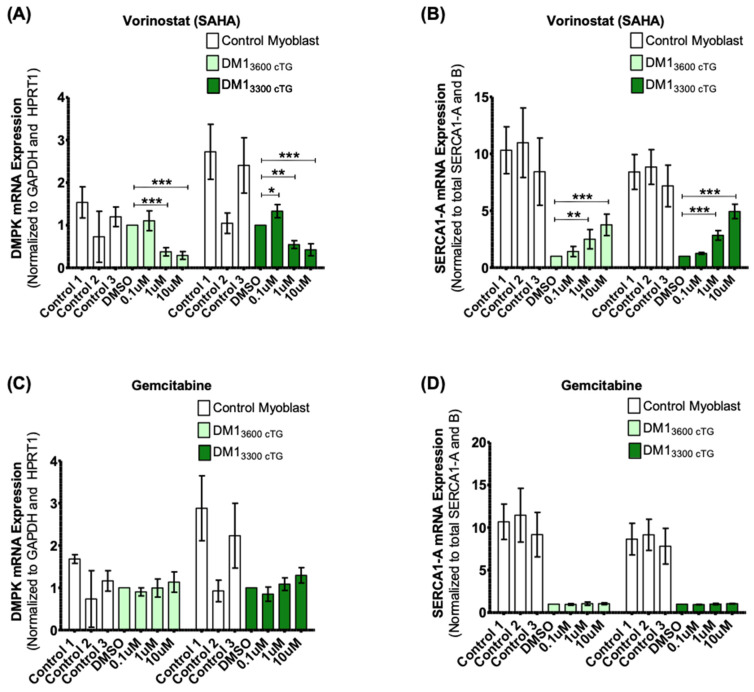
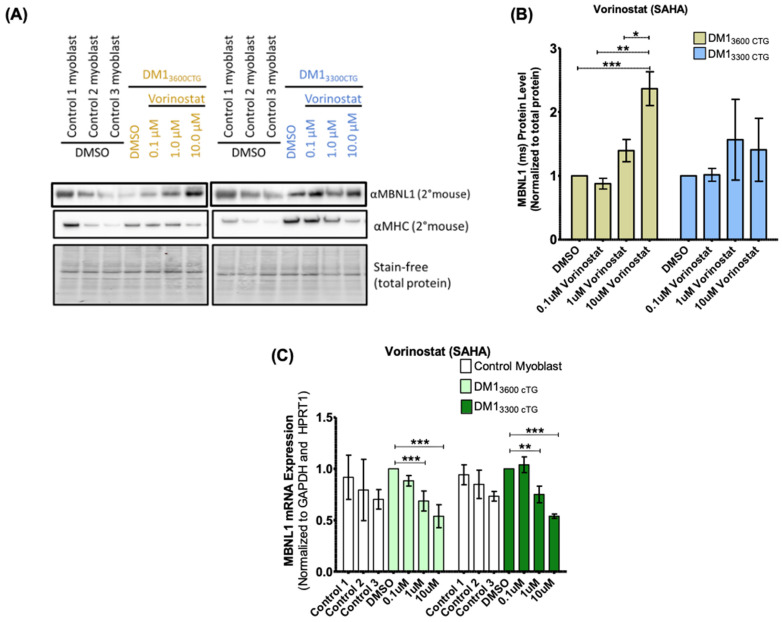
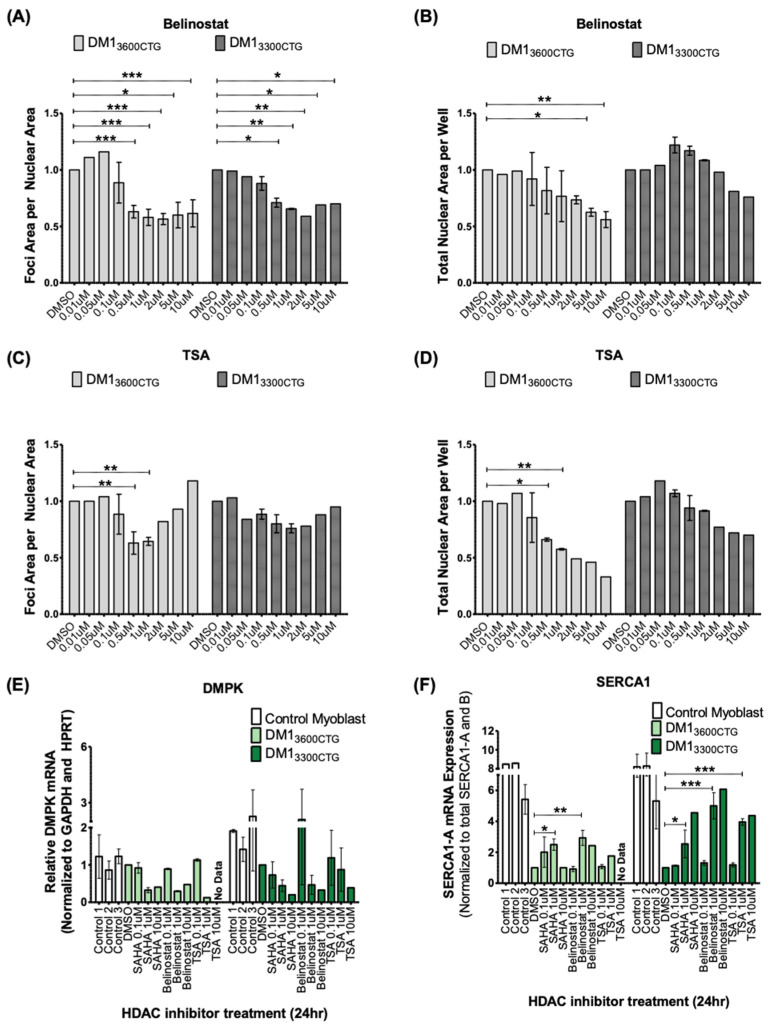
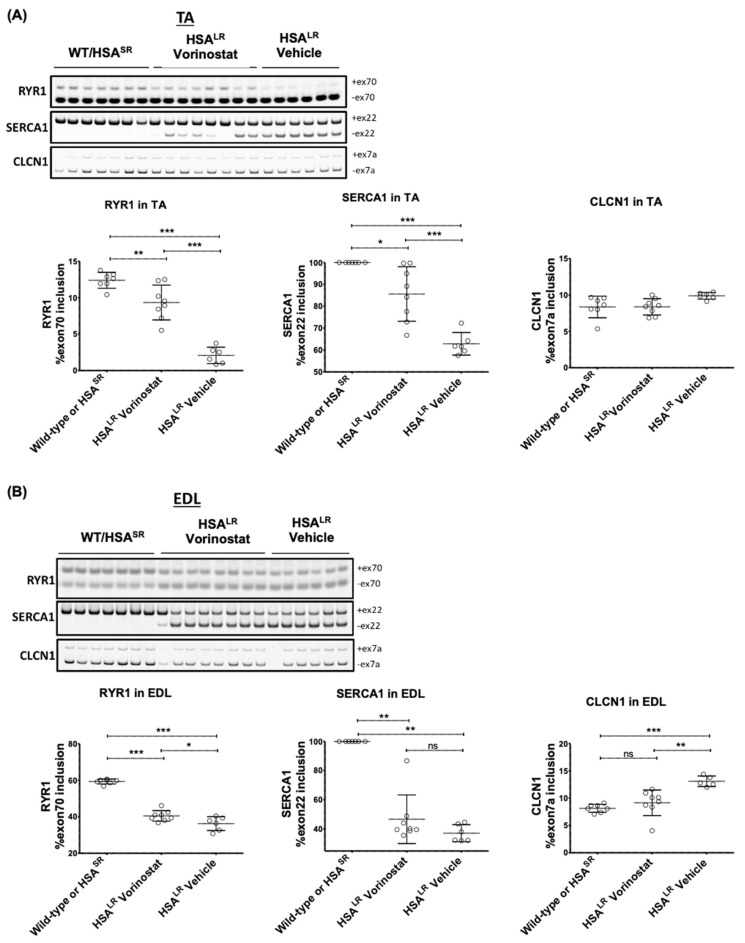
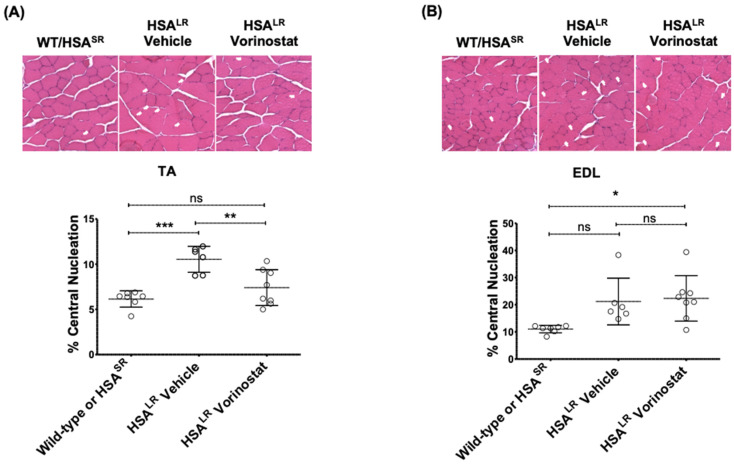
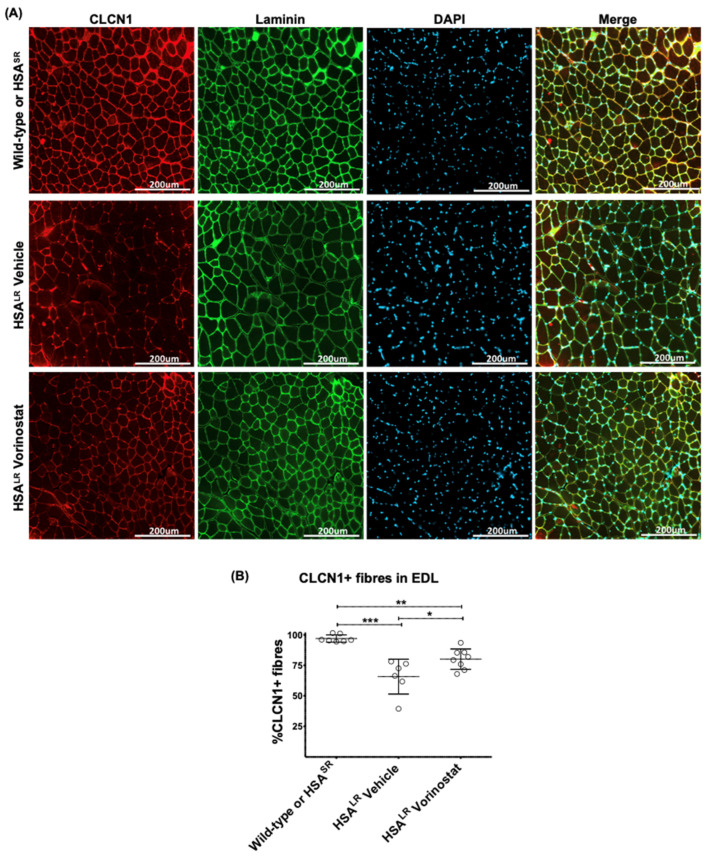
Myotonic dystrophy type 1 (DM1), the most common form of adult muscular dystrophy, is caused by an abnormal expansion of CTG repeats in the 3' untranslated region of the dystrophia myotonica protein kinase (DMPK) gene. The expanded repeats of the DMPK mRNA form hairpin structures in vitro, which cause misregulation and/or sequestration of proteins including the splicing regulator muscleblind-like 1 (MBNL1). In turn, misregulation and sequestration of such proteins result in the aberrant alternative splicing of diverse mRNAs and underlie, at least in part, DM1 pathogenesis. It has been previously shown that disaggregating RNA foci repletes free MBNL1, rescues DM1 spliceopathy, and alleviates associated symptoms such as myotonia. Using an FDA-approved drug library, we have screened for a reduction of CUG foci in patient muscle cells and identified the HDAC inhibitor, vorinostat, as an inhibitor of foci formation; SERCA1 (sarcoplasmic/endoplasmic reticulum Ca2+-ATPase) spliceopathy was also improved by vorinostat treatment. Vorinostat treatment in a mouse model of DM1 (human skeletal actin-long repeat; HSALR) improved several spliceopathies, reduced muscle central nucleation, and restored chloride channel levels at the sarcolemma. Our in vitro and in vivo evidence showing amelioration of several DM1 disease markers marks vorinostat as a promising novel DM1 therapy.
Keywords: myotonic dystrophy type 1; new treatment; rare diseases; vorinostat.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Suominen T., Bachinski L.L., Auvinen S., Hackman P., Baggerly K.A., Angelini C., Peltonen L., Krahe R., Udd B. Population frequency of myotonic dystrophy: Higher than expected frequency of myotonic dystrophy type 2 (DM2) mutation in Finland. Eur. J. Hum. Genet. 2011;19:776–782. doi: 10.1038/ejhg.2011.23. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
