

Fibrosis: Types, Effects, Markers, Mechanisms for Disease Progression, and Its Relation with Oxidative Stress, Immunity, and Inflammation
- PMID: 36835428
- PMCID: PMC9963026
- DOI: 10.3390/ijms24044004
Fibrosis: Types, Effects, Markers, Mechanisms for Disease Progression, and Its Relation with Oxidative Stress, Immunity, and Inflammation
Abstract
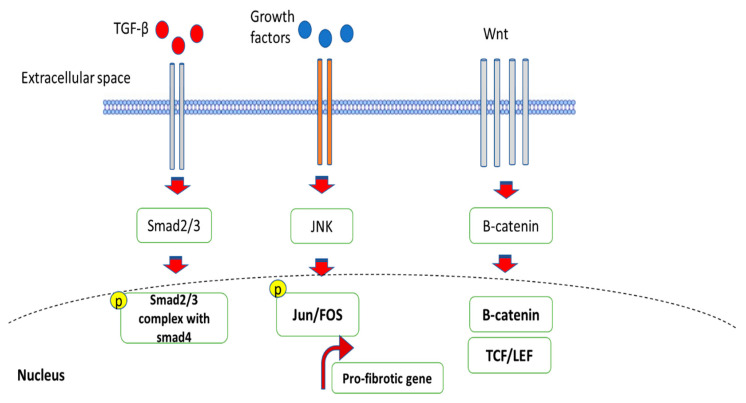
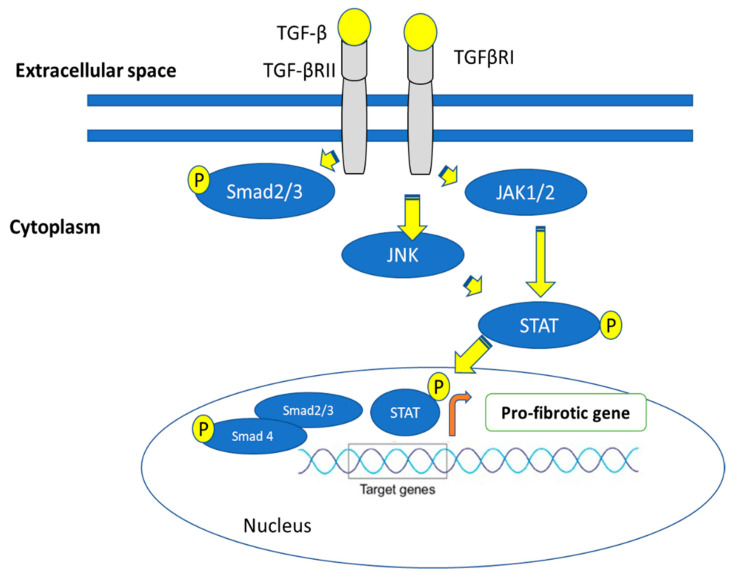
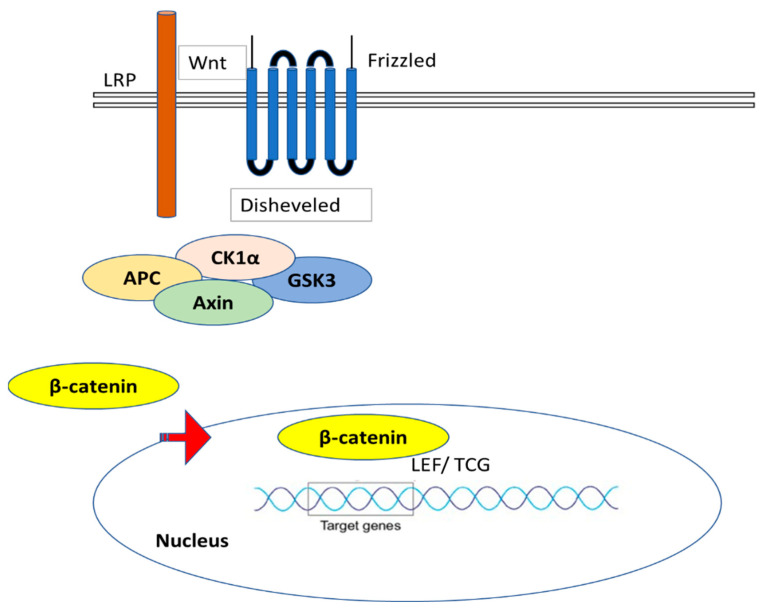
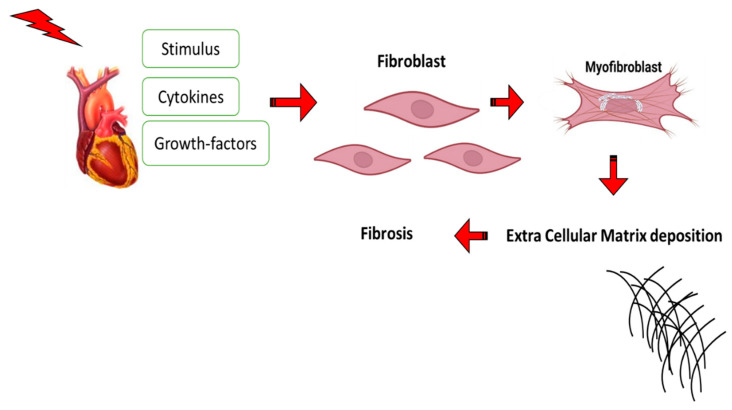
Most chronic inflammatory illnesses include fibrosis as a pathogenic characteristic. Extracellular matrix (ECM) components build up in excess to cause fibrosis or scarring. The fibrotic process finally results in organ malfunction and death if it is severely progressive. Fibrosis affects nearly all tissues of the body. The fibrosis process is associated with chronic inflammation, metabolic homeostasis, and transforming growth factor-β1 (TGF-β1) signaling, where the balance between the oxidant and antioxidant systems appears to be a key modulator in managing these processes. Virtually every organ system, including the lungs, heart, kidney, and liver, can be affected by fibrosis, which is characterized as an excessive accumulation of connective tissue components. Organ malfunction is frequently caused by fibrotic tissue remodeling, which is also frequently linked to high morbidity and mortality. Up to 45% of all fatalities in the industrialized world are caused by fibrosis, which can damage any organ. Long believed to be persistently progressing and irreversible, fibrosis has now been revealed to be a very dynamic process by preclinical models and clinical studies in a variety of organ systems. The pathways from tissue damage to inflammation, fibrosis, and/or malfunction are the main topics of this review. Furthermore, the fibrosis of different organs with their effects was discussed. Finally, we highlight many of the principal mechanisms of fibrosis. These pathways could be considered as promising targets for the development of potential therapies for a variety of important human diseases.
Keywords: ECM; TGF-β; anti-oxidant system; fibrosis; inflammation; organ malfunction.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
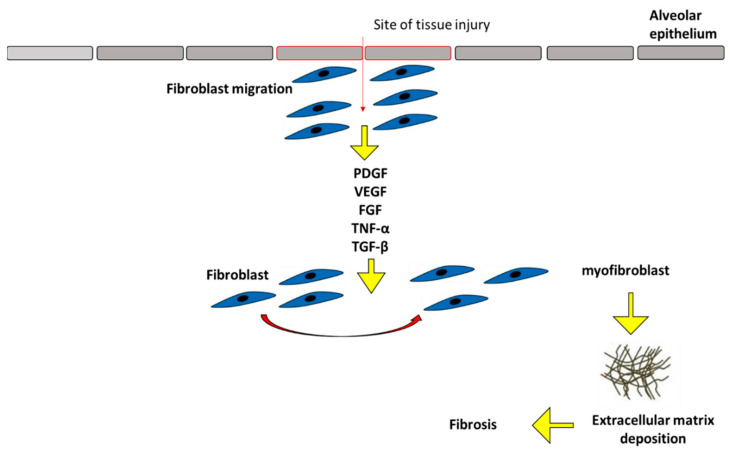
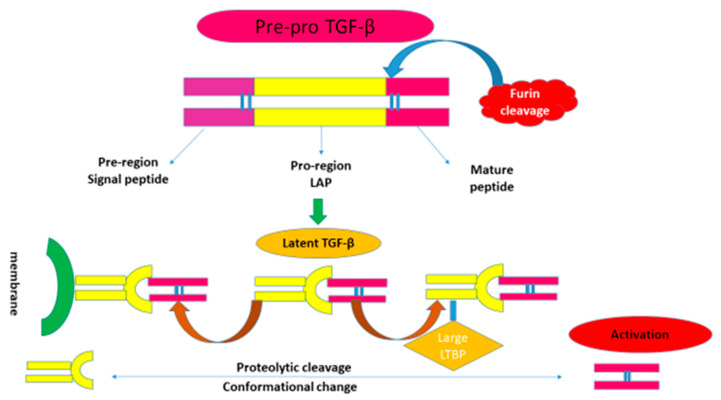
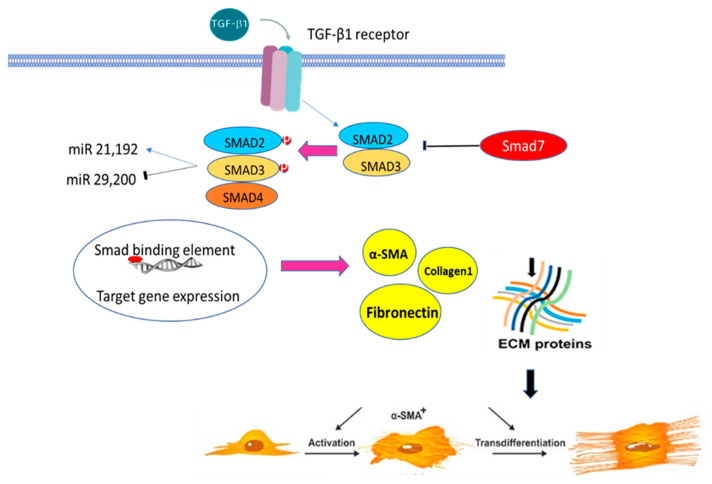
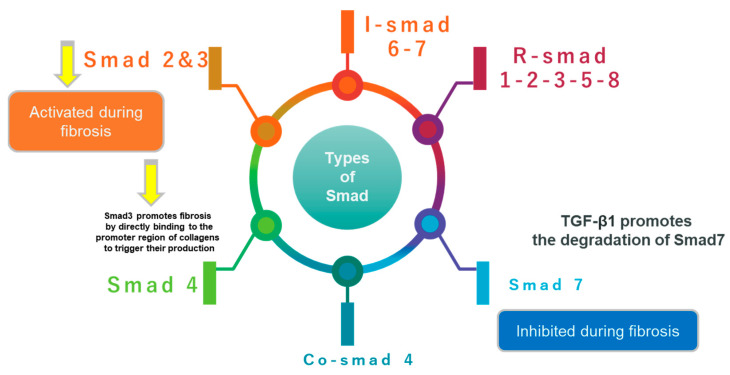
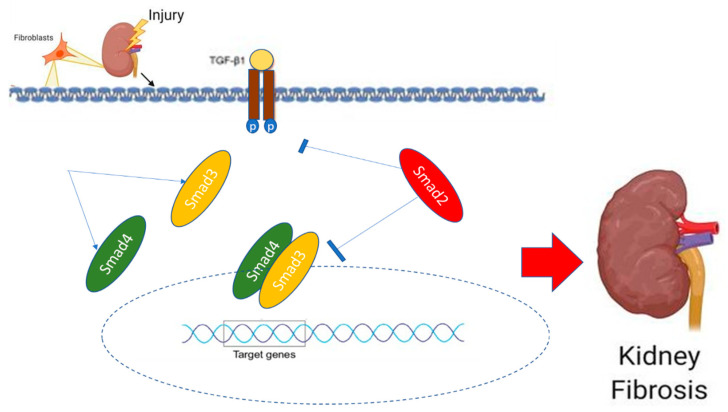
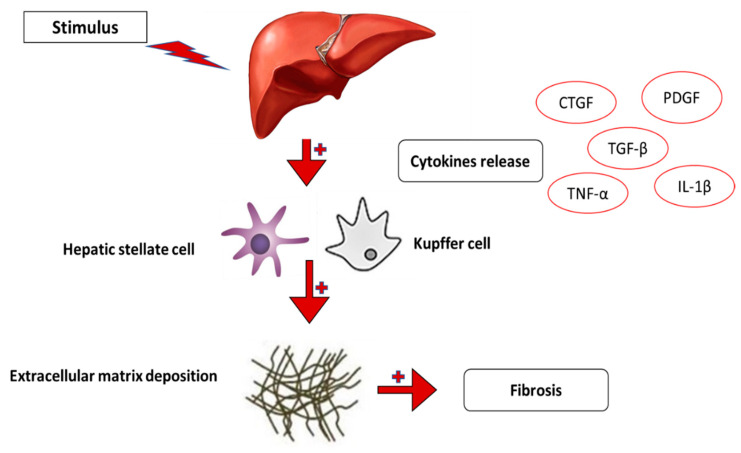
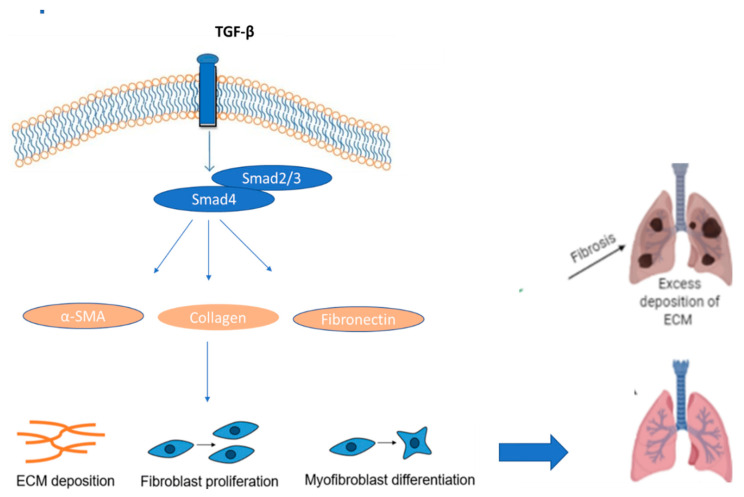
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
