

Cancer Metabolism: Fasting Reset, the Keto-Paradox and Drugs for Undoing
- PMID: 36836124
- PMCID: PMC9960359
- DOI: 10.3390/jcm12041589
Cancer Metabolism: Fasting Reset, the Keto-Paradox and Drugs for Undoing
Abstract
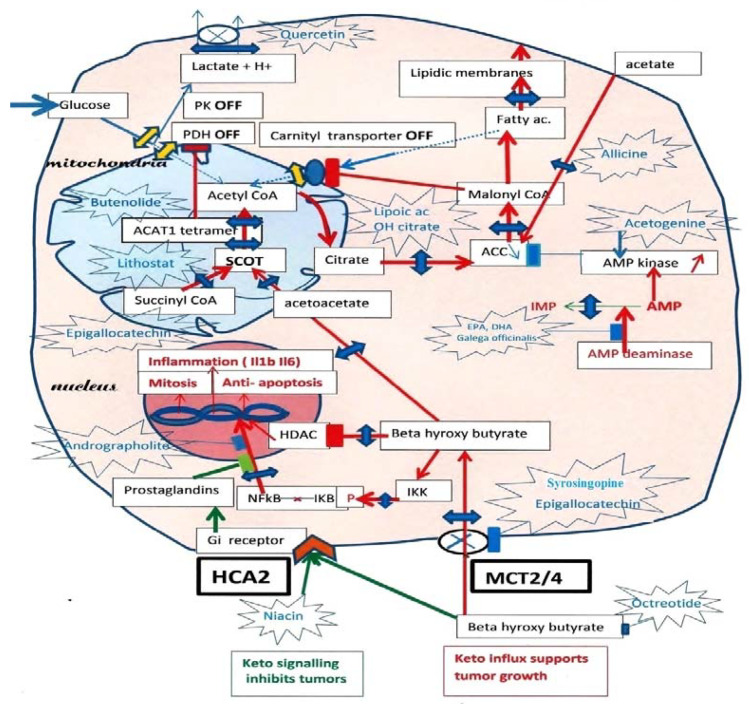
In tumor cells, ketolysis "via" succinyl-CoA: 3-oxoacid-CoAtransferase (SCOT) and acetyl-CoA acetyltransferase 1 (ACAT1) is a major source of mitochondrial acetyl-CoA. Active ACAT1 tetramers stabilize by tyrosine phosphorylation, which facilitates the SCOT reaction and ketolysis. Tyrosine phosphorylation of pyruvate kinase PK M2 has the opposite effect, stabilizing inactive dimers, while pyruvate dehydrogenase (PDH), which is already inhibited by phosphorylation, is acetylated by ACAT1 and is doubly locked. This closes the glycolytic supply of acetyl-CoA. In addition, since tumor cells must synthesize fatty acids to create new membranes, they automatically turn off the degradation of fatty acids into acetyl-CoA ("via" the malonyl-CoA brake for the fatty acid carnityl transporter). Thus, inhibiting SCOT the specific ketolytic enzyme and ACAT1 should hold back tumor progression. However, tumor cells are still able to take up external acetate and convert it into acetyl-CoA in their cytosol "via" an acetyl-CoA synthetase, which feeds the lipogenic pathway; additionally, inhibiting this enzyme would make it difficult for tumor cells to form new lipid membrane and survive.
Keywords: SCOT-ACAT1 inhibitions; acetohydroxamates; butenolides; karrikins; siderophores; tumor ketolysis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Mazurek S., Eigenbrodt E. The tumor metabolome. Anticancer Res. 2003;23:1149–1154. - PubMed
-
- Eigenbrodt E., Gerbracht U., Mazurek S., Presek P., Friis R. Carbohydrate metabolism and neoplasia: New Perspectives for diagnosis and therapy. In: Prestlow T.G., Prestlow T.P., editors. Biochemical and Molecular Aspects of Selected Cancers. Volume 2. Academic Press; Cambridge, MA, USA: 1994. pp. 311–385.
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous
