

The Mechanism of Hyperglycemia-Induced Renal Cell Injury in Diabetic Nephropathy Disease: An Update
- PMID: 36836895
- PMCID: PMC9967500
- DOI: 10.3390/life13020539
The Mechanism of Hyperglycemia-Induced Renal Cell Injury in Diabetic Nephropathy Disease: An Update
Abstract
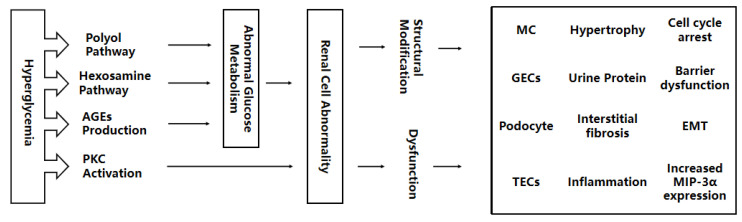
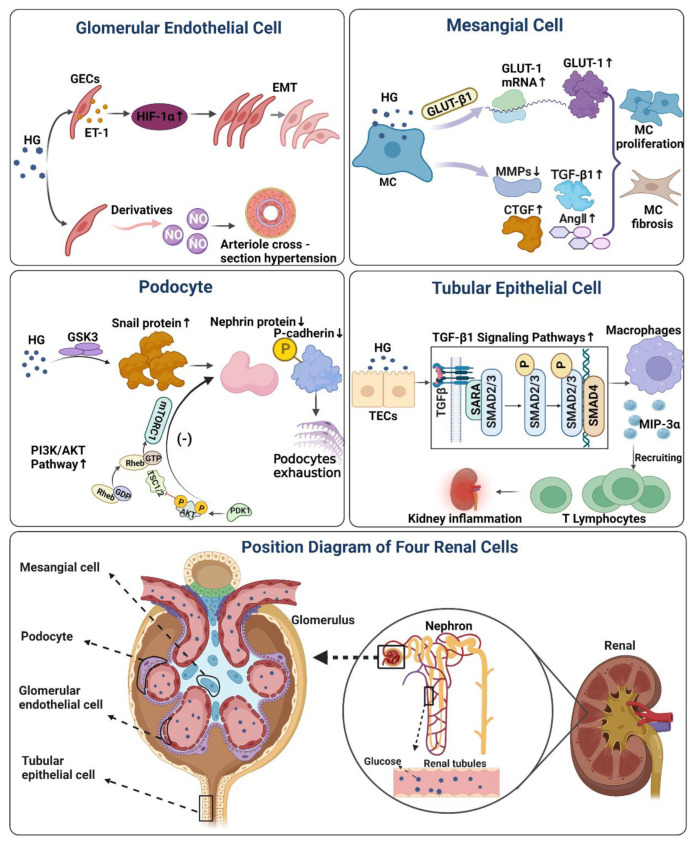
Diabetic Nephropathy (DN) is a serious complication of type I and II diabetes. It develops from the initial microproteinuria to end-stage renal failure. The main initiator for DN is chronic hyperglycemia. Hyperglycemia (HG) can stimulate the resident and non-resident renal cells to produce humoral mediators and cytokines that can lead to functional and phenotypic changes in renal cells and tissues, interference with cell growth, interacting proteins, advanced glycation end products (AGEs), etc., ultimately resulting in glomerular and tubular damage and the onset of kidney disease. Therefore, poor blood glucose control is a particularly important risk factor for the development of DN. In this paper, the types and mechanisms of DN cell damage are classified and summarized by reviewing the related literature concerning the effect of hyperglycemia on the development of DN. At the cellular level, we summarize the mechanisms and effects of renal damage by hyperglycemia. This is expected to provide therapeutic ideas and inspiration for further studies on the treatment of patients with DN.
Keywords: diabetic nephropathy; hyperglycemia; mechanism; renal cell injury.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Thomas S., Karalliedde J. Diabetic nephropathy. Medicine. 2015;43:20–25. doi: 10.1016/j.mpmed.2014.10.007. - DOI
Publication types
LinkOut - more resources
Full Text Sources
