

Femtosecond Time-Resolved Observation of Relaxation and Wave Packet Dynamics of the S1 State in Electronically Excited o-Fluoroaniline
- PMID: 36838988
- PMCID: PMC9965681
- DOI: 10.3390/molecules28041999
Femtosecond Time-Resolved Observation of Relaxation and Wave Packet Dynamics of the S1 State in Electronically Excited o-Fluoroaniline
Abstract
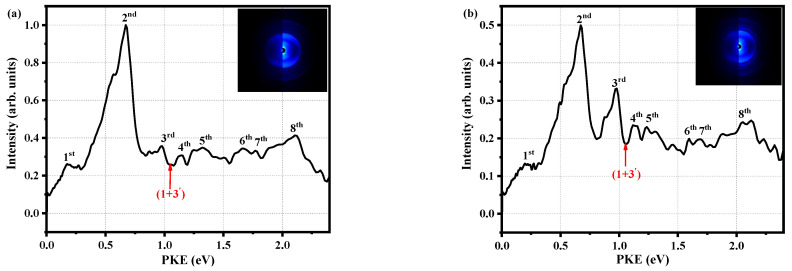
Quantum beat frequency is the basis for understanding interference effects and vibrational wave packet dynamics and has important applications. Using femtosecond time-resolved mass spectrometry and femtosecond time-resolved photoelectron image combined with theoretical calculations, we study the electronic excited-state relaxation of o-fluoraniline molecule and the time-dependent evolution of vibrational wave packets between different eigenstates. After the molecule absorbs a photon of 288.3 nm and is excited to the S1 state, intramolecular vibrational redistribution first occurs on the time scale τ1 = 349 fs, and then the transition to the triplet state occurs through the intersystem crossing on the time scale τ2 = 583 ps, and finally, the triplet state occurs decays slowly through the time scale τ3 = 2074 ps. We find the intramolecular vibrational redistribution is caused by the 00, 10b1 and 16a1 vibrational modes of the Sl state origin. That is, the 288.3 nm femtosecond laser excites the molecule to the S1 state, and the continuous flow of the vibrational wave packet prepares a coherent superposition state of three vibrational modes. Through extracting the oscillation of different peak intensities in the photoelectron spectrum, we observe reversible changes caused by mutual interference of the S1 00, S1 10b1 and S1 16a1 states when the wave packets flow. When the pump pulse is 280 nm, the beat frequency disappears completely. This is explained in terms of increases in the vibrational field density and characteristic period of oscillation, and statistical averaging makes the quantum effect smooth and indistinguishable. In addition, the Rydberg component of the S1 state is more clearly resolved by combining experiment and theory.
Keywords: o-fluoraniline; photoelectron image; quantum beat frequency; relaxation dynamic; wave packet dynamic.
Conflict of interest statement
The authors have no conflict to declare.
Figures









References
-
- Kuthirummal N., Weber P.M. Rydberg states: Sensitive probes of molecular structure. Chem. Phys. Lett. 2003;378:647–653. doi: 10.1016/j.cplett.2003.08.002. - DOI
-
- Cheng X.X., Zhang Y., Deb S., Minitti M.P., Gao Y., Jónssonad H.P., Weber M. Ultrafast structural dynamics in Rydberg excited N,N,N′,N′-tetramethylethylenediamine: Conformation dependent electron lone pair interaction and charge delocalization. Chem. Sci. 2014;5:4394–4403. doi: 10.1039/C4SC01646G. - DOI
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
