

NRF2 activation induces NADH-reductive stress, providing a metabolic vulnerability in lung cancer
- PMID: 36841242
- PMCID: PMC9998367
- DOI: 10.1016/j.cmet.2023.01.012
NRF2 activation induces NADH-reductive stress, providing a metabolic vulnerability in lung cancer
Erratum in
-
NRF2 activation induces NADH-reductive stress, providing a metabolic vulnerability in lung cancer.Cell Metab. 2023 Apr 4;35(4):722. doi: 10.1016/j.cmet.2023.03.011. Cell Metab. 2023. PMID: 37019082 Free PMC article. No abstract available.
Abstract
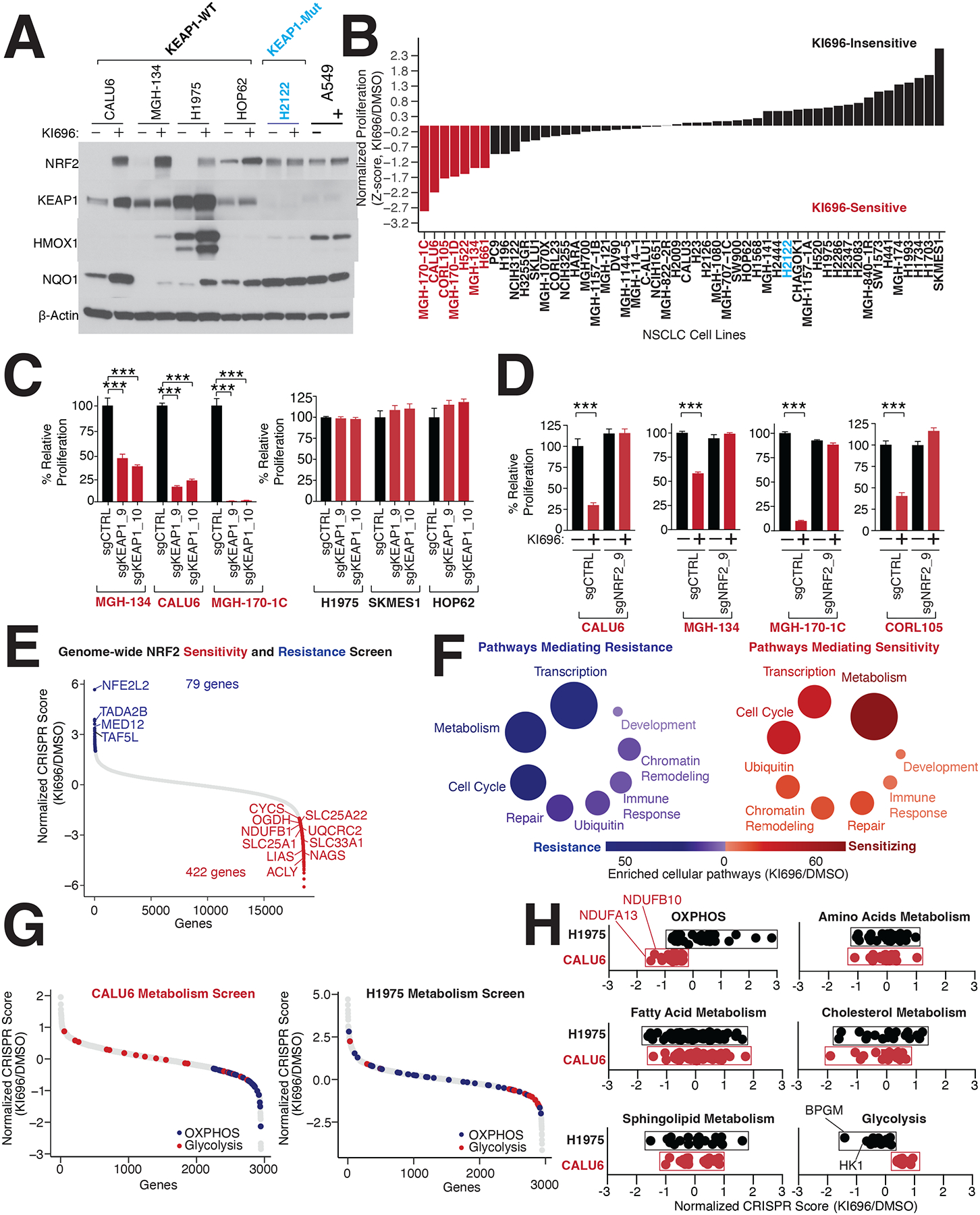
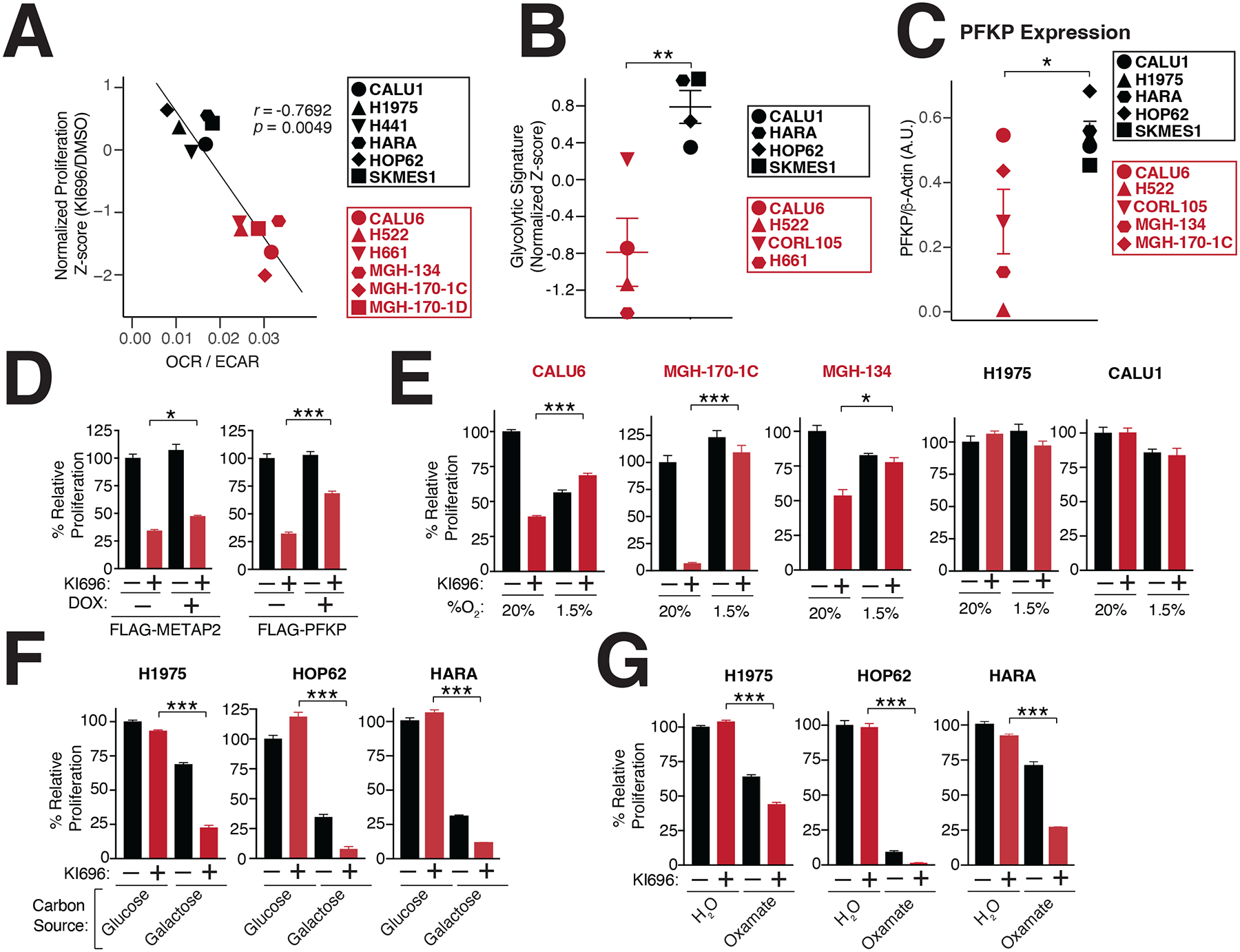
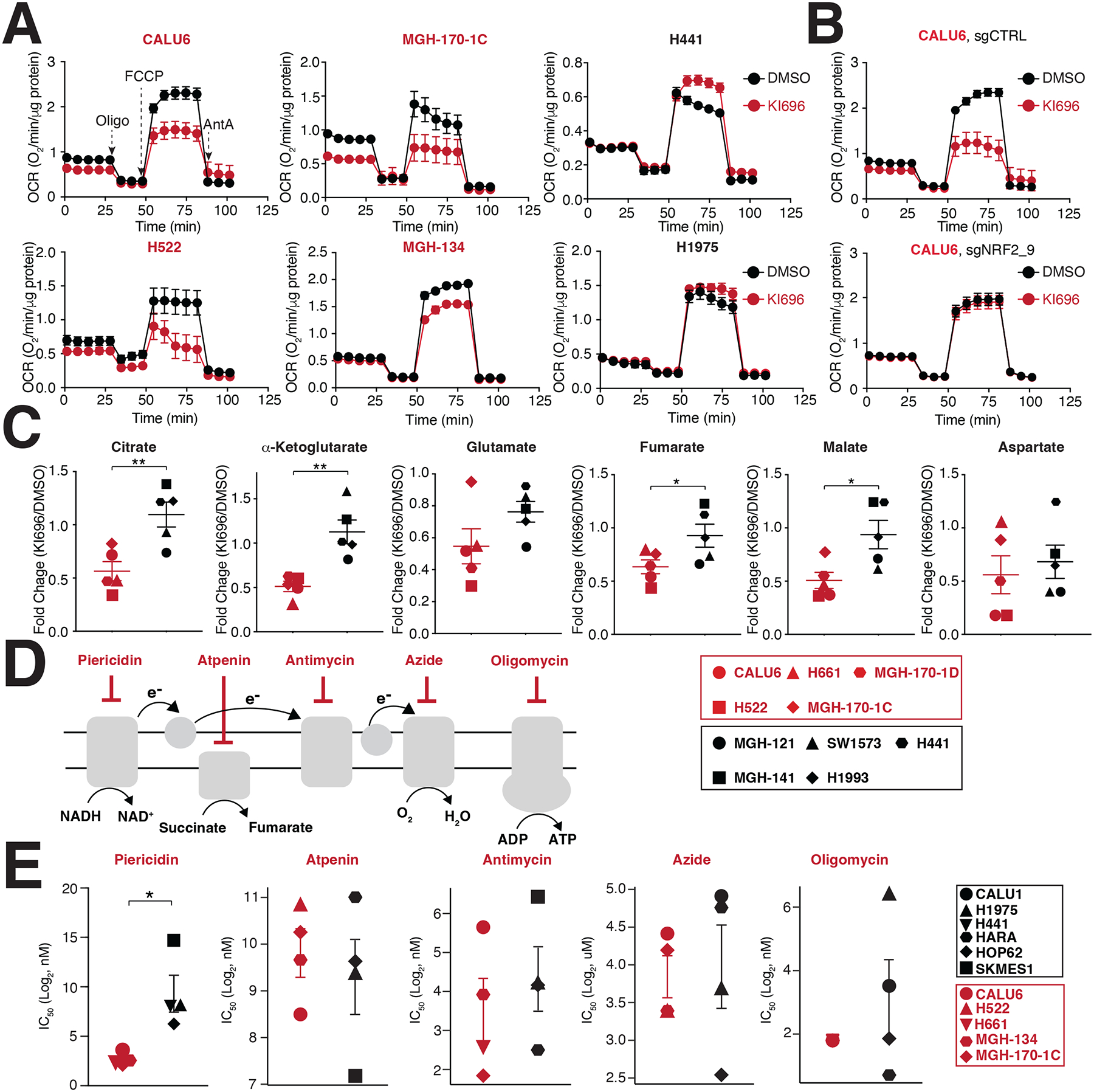
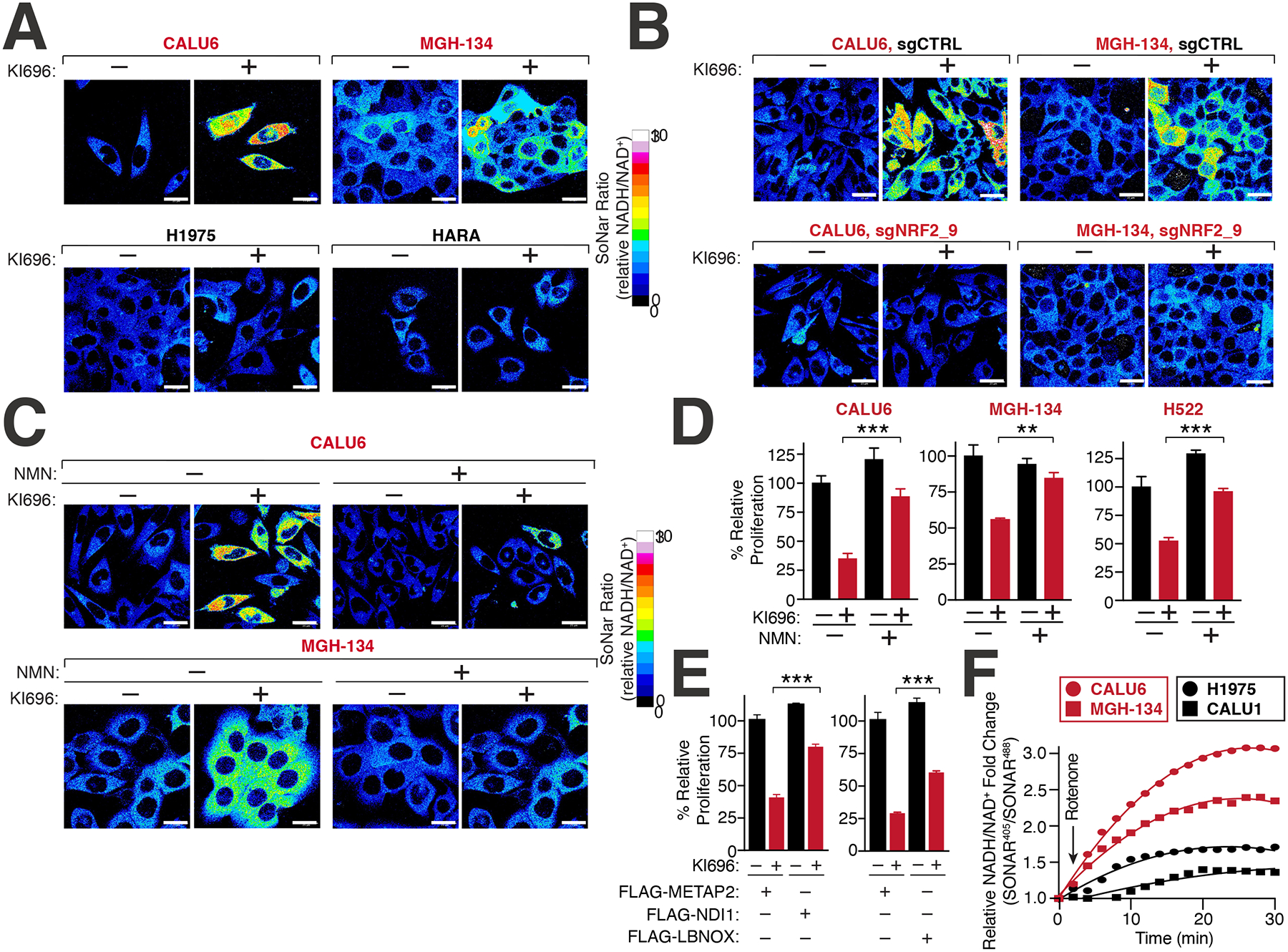
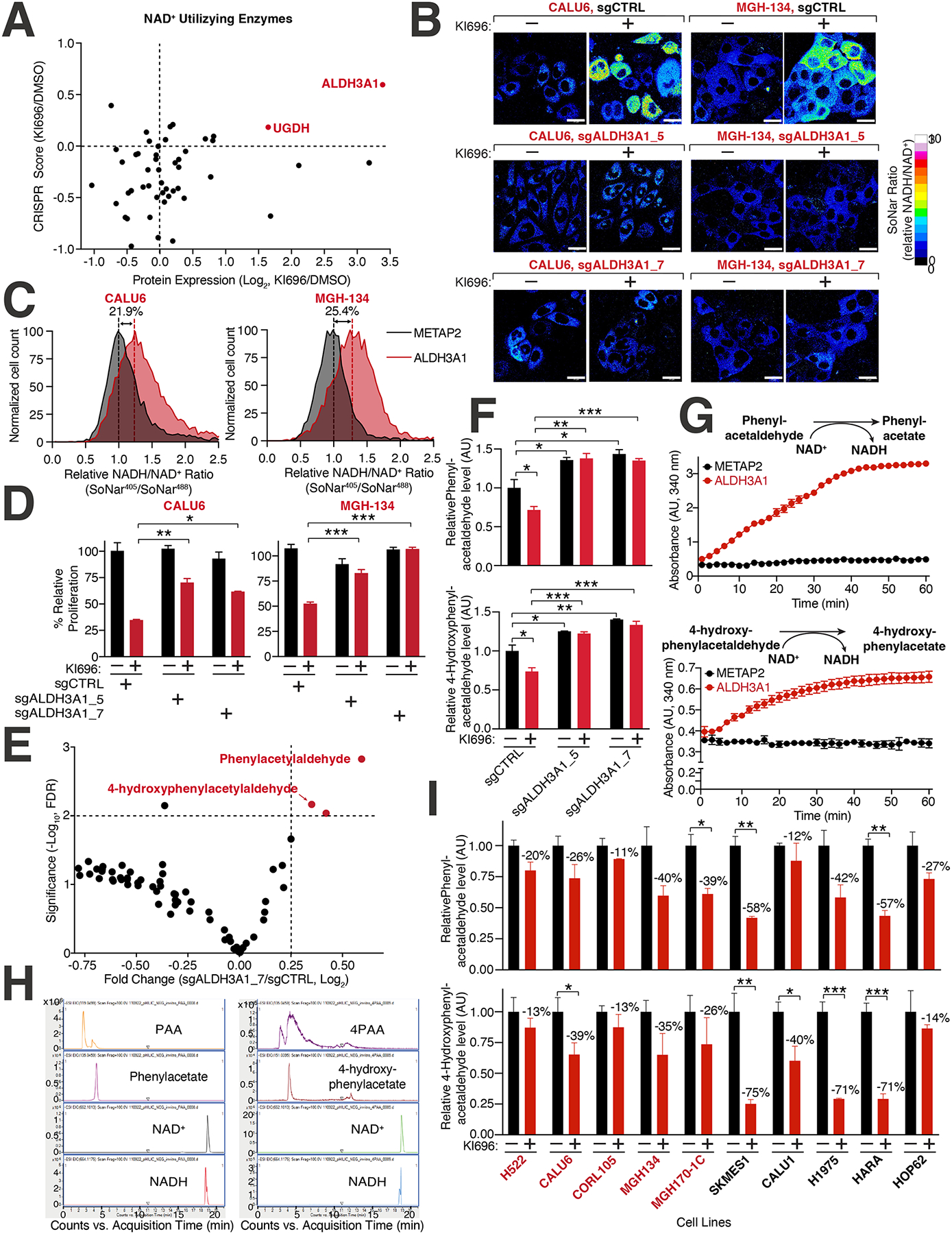
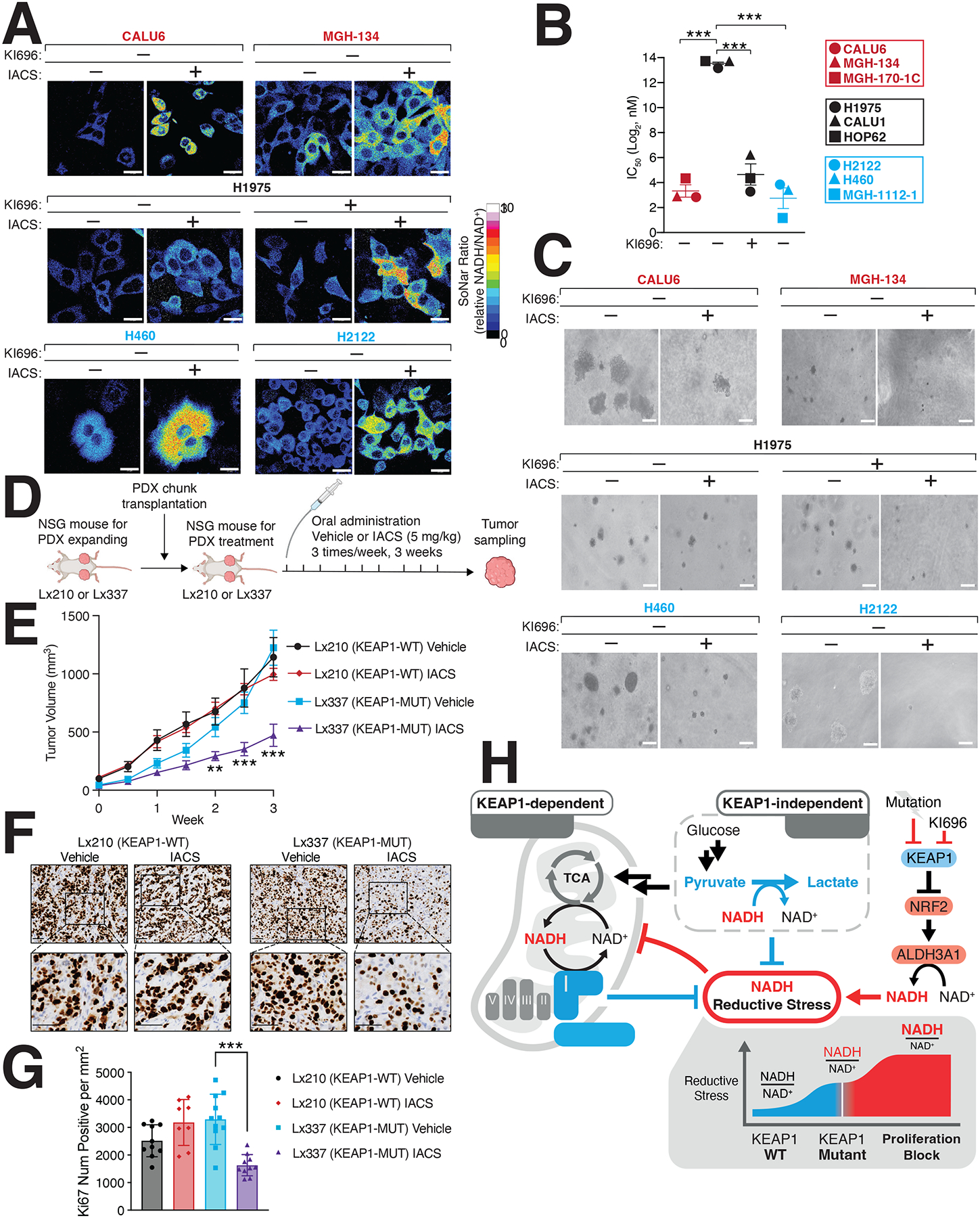
Multiple cancers regulate oxidative stress by activating the transcription factor NRF2 through mutation of its negative regulator, KEAP1. NRF2 has been studied extensively in KEAP1-mutant cancers; however, the role of this pathway in cancers with wild-type KEAP1 remains poorly understood. To answer this question, we induced NRF2 via pharmacological inactivation of KEAP1 in a panel of 50+ non-small cell lung cancer cell lines. Unexpectedly, marked decreases in viability were observed in >13% of the cell lines-an effect that was rescued by NRF2 ablation. Genome-wide and targeted CRISPR screens revealed that NRF2 induces NADH-reductive stress, through the upregulation of the NAD+-consuming enzyme ALDH3A1. Leveraging these findings, we show that cells treated with KEAP1 inhibitors or those with endogenous KEAP1 mutations are selectively vulnerable to Complex I inhibition, which impairs NADH oxidation capacity and potentiates reductive stress. Thus, we identify reductive stress as a metabolic vulnerability in NRF2-activated lung cancers.
Keywords: NADH/NAD(+); NRF2-KEAP1 pathway; functional genomic; non-small cell lung cancer; oxidative phosphorylation; reductive stress.
Copyright © 2023 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests L.B-P is a founder, consultant, and holds privately held equity in Scorpion Therapeutics. D.E.F. has a financial interest in Soltego, a company developing salt-inducible kinase inhibitors for topical skin-darkening treatments that might be used for a broad set of human applications. The interests of L.B-P and D.E.F. were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict-of-interest policies. M.C.H. is on the SAB and holds equity in Alixia Therapeutics and is on the SAB of Minovia. T. P. is on the SAB of Faeth Therapeutics and consults for Dracen Pharmaceuticals. A.N.H receives research funding from Amgen, Blueprint Medicines, BridgeBio, Bristol-Myers Sqiub, C4 Therapeutics, Eli Lilly, Novartis, Nuvalent, Pfizer, Roche/Genentech, and Scorpion Therapeutics, and consults for Engine Biosciences, Nuvalent, TigaTx, and Tolremo Therapeutics.
Figures
References
-
- Cantor JR & Sabatini DM (2012). Cancer cell metabolism: one hallmark, many faces. Cancer Discov 2, 881–898, doi:10.1158/2159-8290.CD-12-0345. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U01 CA267827/CA/NCI NIH HHS/United States
- R01 AR072304/AR/NIAMS NIH HHS/United States
- R01 CA235412/CA/NCI NIH HHS/United States
- P50 CA127003/CA/NCI NIH HHS/United States
- R01 GM128448/GM/NIGMS NIH HHS/United States
- R00 CA215249/CA/NCI NIH HHS/United States
- P01 CA163222/CA/NCI NIH HHS/United States
- R01 CA273461/CA/NCI NIH HHS/United States
- R37 CA260062/CA/NCI NIH HHS/United States
- R01 CA227649/CA/NCI NIH HHS/United States
- R21 CA256082/CA/NCI NIH HHS/United States
- R01 CA219670/CA/NCI NIH HHS/United States
- R01 CA222871/CA/NCI NIH HHS/United States
- R01 AR043369/AR/NIAMS NIH HHS/United States
- R37 CA222504/CA/NCI NIH HHS/United States
- R01 CA215498/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
